THE OMICRON VARIANT BREAKS THE EVOLUTIONARY LINEAGEOF SARS-COV2 VARIANTSJean Claude Perez 1 1 PhD Maths and Computer Science Bordeaux University; Retired (IBM European Research center on Artificial Intelligence Montpellier France); Bordeaux metropole, France.2 PhD; EMBL Heidelberg alumni, Meyerhofstraße 1, 69117 Heidelberg, Germany.3 Fondation Luc Montagnier Quai Gustave-Ador 62 1207 Genève, Switzerland. |
|
||
|
|
|||
|
Received 4 November 2021 Accepted 15 December 2021 Published 31 December 2021 Corresponding Author Jean
Claude Perez, jeanclaudepere2@gmail.com DOI 10.29121/granthaalayah.v9.i12.2021.4418 Funding:
This
research received no specific grant from any funding agency in the public,
commercial, or not-for-profit sectors. Copyright:
© 2021
The Author(s). This is an open access article distributed under the terms of
the Creative Commons Attribution License, which permits unrestricted use, distribution,
and reproduction in any medium, provided the original author and source are
credited.
|
ABSTRACT |
|
|
|
We
analyzed 15 genomes and Spikes of the new OMICRON variant, on the one hand 7
from the very first 21K lineage (South Africa, USA, Belgium, Canada), on the
other hand 8 from the later second sister-clade 21L (USA, Switzerland, UK). We
applied, at the scale of the whole genome and the spike gene, the
biomathematics method of Fibonacci meta-structure fractal analysis applied to
the UA / CG proportions. There
appears a total rupture of this variant with respect to all the previous
variants, and a strong differentiation between these 2 OMICRON lines. We have
evidenced the RUPTURE of OMICRON with respect to ALL the previous variants:
D614G, ALPHA, BETA, GAMMA, DELTA. In
particular, we suggest that the mRNA stabilizing secondary structure
("hairpin" conformation) in the spike of all variants is degraded
in OMICRON, probably making its mRNA more fragile. The
loss of long-range fractal meta-structures in the OMICRON spike gene are in
line with common knowledge on the mechanisms of epidemic ending, involving
recombination of heavily mutated RNA fragments of the virus, with the
possible inference of a distinct helper virus. This would indicate that the
SARS-CoV2 is under very strong evolutionary pressure, possibly marking the
end of the pandemic. Remarkably,
it is observed that the density of OMICRON mutations in the SPIKE PRION
region is more than 8 times that of the rest of the Spike protein. This high
density of mutations of the Prion region in OMICRON appears to completely
suppress this possible Prion function, unlike the Spikes of the various
variants and mRNAs vaccines where this Prion function is observed. |
|
||
|
Keywords: Covid-19,
SARS-CoV-2, Epidemic Ending, Variant Omicron, Collective Immunity, Genome
Order, Fractal Metastructures, Prion Protein Function, Fibonacci “Science doesn't tell us where we're going - that's the role of art -;
it tells us where we are.” Alan
TURING ADDENDUM by Pr Luc Montagnier "A big step towards understanding Nature. Your work gives us hope that we will soon emerge from the nightmare
where the Cretinoid branch of Humanity plunges us” Luc Montagnier |
|
||
1. INTRODUCTION
Throughout this pandemic the consensus between TV studio allowed scientists and back-office scientists hardly shone. One could even say that the
established consensus was unidirectional behaving like the electronic diodes of the ancestors of our computers. The scientific current could only pass in one direction without contradiction. However, the OMICRON variant, produces a real general consensus: all agree to note and then to affirm that it constitutes a RUPTURE with the preceding ALPHA, BETA, GAMMA and DELTA variants.
Faced with this accelerated evolution of the new SARS-CoV2 genome variant, the little-known fractal-based biomathematic analysis methods we have developed could possibly capture essential epidemiological characteristics of this new variant, that is expected to become predominant in the world. With this analysis we intend to provide a mathematical measure to put the two-fold question of transmission and pathogenicity of the OMICRON variant in perspective with rationally established but forgotten knowledge on the dynamics of epidemics leading to their ending.
In two articles published in 2021 Perez (2021a) and Perez (2021b) , we showed how the FIBONACCI fractal analysis method makes it possible to measure the progressive adaptation of successive variant genomes to the human host. Thus, we demonstrated how at the double levels of the spike and of the whole genome, Fibonacci meta-structures in UA / CG proportions of covering the entire genome sequence were permanently reinforced between the original Wuhan strain then the 3 successive worldwide variants D614G, ALPHA, and DELTA.
This genome analysis method makes it possible to measure the consistency and structural homogeneity of a genome. For example, in a previous work Perez (2018) we demonstrated how the small human mitochondrial mtDNA genome lost its digital Fibonacci metastructure during mutations associated with various cancers.
2. METHODS
2.1. Computing FIBONACCI metastructures
Consider the sequence of Fibonacci numbers:
0 1 1 2 3 5 8 13 21 34 55 89 144 233 377 610 987 1597 2584 4181 6765 10946 17711 28657 46368 75025 121393 196418 317811 514229 832040 1346269 2178309 3524578 5702887...
Example of the SPIKE from WUHAN reference genome, this mRNA SPIKE is 3822 UACG bases in length.
Recall WUHAN reference https://www.ncbi.nlm.nih.gov/nuccore/NC_045512
Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome NCBI Reference Sequence: NC_045512.2
The longest Fibonacci structures would therefore measure 2584 bases. When looking for such structures, the first one found is at location 1200:
therefore, covering the bases situated between 1201 and 3784 (1200 + 2584).
These 2584 bases are broken down respectively into:
1597 bases UA
and 987 bases CG
Here are the first 20 basics that the reader can easily check:
SPIKREF [1200+¼20]
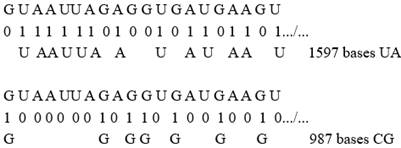
The Spike analysis of this Wuhan-Hu-1 reference genome reports 63 metastructures of this type if we close the sequence on itself (as in mtDNA or bacteria) and 7 metastructures if we consider the mRNA sequence in its unclosed linear form, as will be the case throughout this study.
2.2. Analysis of reference OMICRON variant strains:
We analyzed seven OMICRON variant genomes and spikes mRNA sequences.
The three original South African sequences received from the teams of Pr Tulio de Oliveira, Dr Penny Moore and Dr Cathrine Scheepers (HIV & SARS-CoV-2 Virology Section, Centre for HIV & STI’s, National Institute for Communicable Diseases (NICD), a Division e analysedof the NHLS SAMRC Antibody Immunity Research Unit (AIRU), 1 Modderfontein Road, Sandringham, 2131, South Africa).
The first California sequence was received from Professor Charles Chiu, M.D./Ph.D., Laboratory Medicine and Medicine / Infectious Diseases Director, UCSF-Abbott Viral Diagnostics and Discovery Center Associate Director, UCSF Clinical Microbiology Laboratory UCSF School of Medicine.
Three other sequences were obtained with BLAST using a selection area of the 114 nucleotides PRION region Tetz and Tetz (2020) with their 8 amino acids mutations in OMICRON; see https://covariants.org/variants/21K.Omicron

Then finally, we analyzed seven OMICRON variants sequences:
· 3 from South Afrika
· 1 -USA-California (first case detected)
· 3 from NCBI (prion primer 38 AA with the 8 OMICRON mutations)
The PRION region described in the work of Tetz and Tetz (2020) are located between amino acids 473-510 of the Spike protein.
The 8 mutated amino acids between the reference SARS-CoV2 Spike and OMICRON variant Spike are S477N, T478K, E484A, Q493K, G496S, Q498R, N501Y and Y505H.
We must recall here that amino acids Q and N (Glutamate and Aspargine are the main amino acids favoring PRION properties (Q: CAA CAG; N: AAT AAC).



OMICRONSA1 29816 bases hCoV-19/South Africa/NICD-N21675/2021
OMICRONSA2 29841 bases hCoV-19/South Africa/NICD-N21668/2021
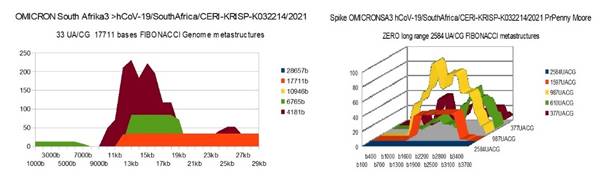
OMICRONSA3 29760 bases hCoV-19/SouthAfrica/CERI-KRISP-K032214/2021
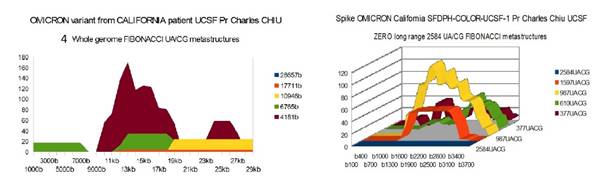
OMICRONCAL 29858 bases SFDPH-COLOR-UCSF-1 Pr CHARLES CHIU
OMICRONBEL 29684 bases SARS-CoV-2/human/BEL/rega-20174/2021 Sequence ID: OL672836.1
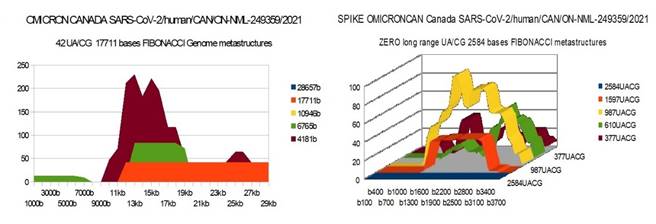
OMICRONCAN 29673 bases SARS-CoV-2/human/CAN/ON-NML-249359/2021 Sequence ID: OL677199.1
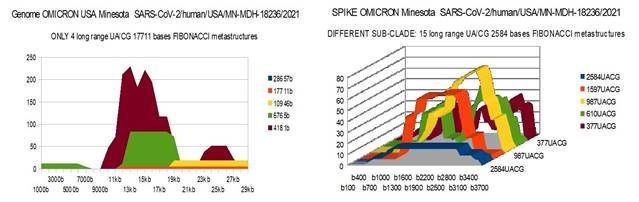
OMICRONMIN 29337 bases SARS-CoV-2/human/USA/MN-MDH-18236/2021 Sequence ID: OL698718.1
3. RESULTS
|
Table 1 Summary table of the number of longest UA / CG FIBONACCI metastructures of Genomes (17711 bases) and Spikes (2584 bases). |
||||
|
Reference |
Long range Genomic 17711 UA/CG metastructures |
Long range Spike 2584 UA/CG metastructures |
Long range Spike « Podium like » 1587 UA/CG metastructures |
Notes |
|
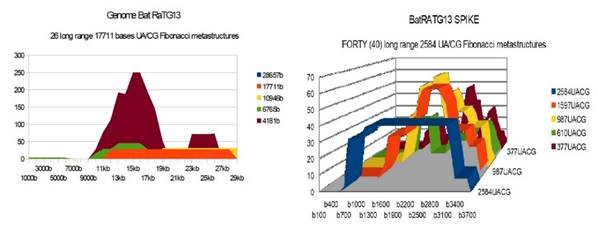
Bat RaTG13 |
26 |
40 |
28 59 31 (a) |
(a) ALL « podium like » 1597 UA/CG metastructures |
|
Initial Wuhan first sequence |
8 |
7 |
37 64 26 |
|
|
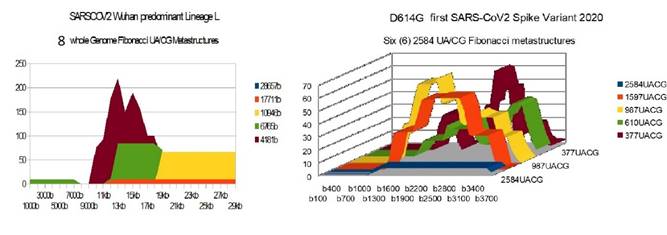
Wuhan Lineage D614G |
8 |
5 |
18 44 26 |
|
|
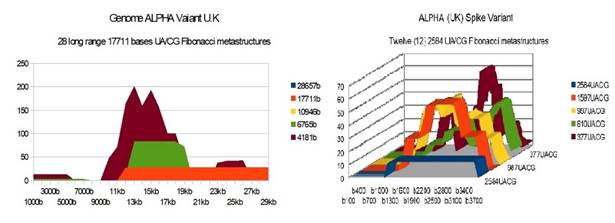
ALPHA UK variant |
28 |
12 |
25 51 26 |
|
|
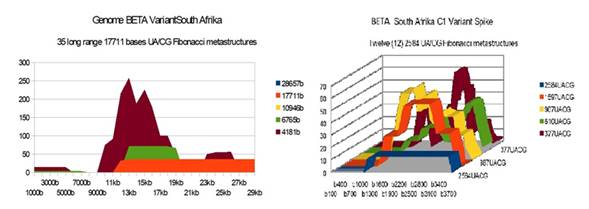
BETA South Afrika |
35 |
12 |
25 51 26 |
|
|
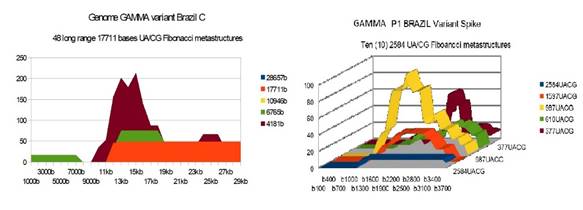
GAMMABrazil |
48 |
10 |
19 34 15 |
|
|
CALifornia variant CAL20C |
5 |
6 |
44 70 26 |
|
|
MINK |
5 |
6 |
41 67 26 |
|
|
MARSEILLE4 |
33 |
5 |
18 44 26 |
|
|
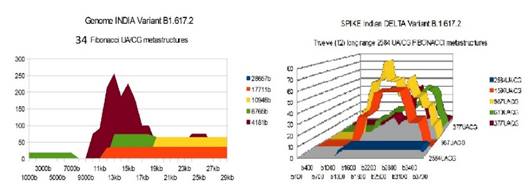
B1.617 INDIA |
53 |
12 |
41 67 26 |
|
|
B.1.617.2 DELTA |
34 |
7 |
30 54 24 |
|
|
OMICRON |
||||
|
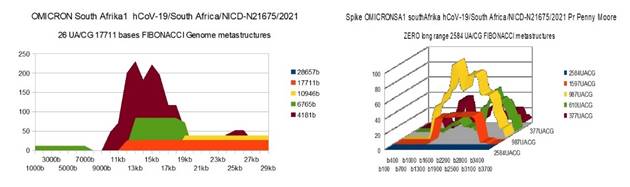
OMICRON SA1 |
26 |
0 (b) |
36 (c) |
(b) ALL OMICRON Spike have ZERO long 2584 UA/CG |
|
(c) The Podium like metastructure is broken |
||||
|
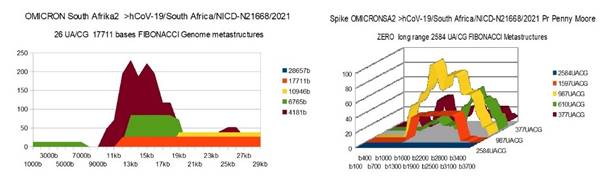
OMICRON SA2 |
26 (d) |
0 |
36 |
(d) OMICRON Variants from South Afrike, Europe and Canada have
genomic 17711 long metastructures near those of DELTA variant |
|
OMICRON SA3 |
33 |
0 |
36 |
|
|
OMICRON CAL |
4 (e) |
0 |
49 |
(e) The 2 first USA OMICRON have a very low number of long range 17711
UA/CG metastructures |
|
OMICRON MIN |
4 |
15 |
58 |
OMICRON Minesota
is from a different OMICRON sub-clade |
|
OMICRON CAN |
42 |
0 |
36 |
|
|
OMICRON BEL |
26 |
0 |
36 |
|
Detailed figures
|
|
|
Figure 1 BatRaTG13
Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 2 Wuhan
lineage L D614G Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 3 ALPHA U.K variant Genome and
Spike Fibonacci UA/CG metastructures |
|
|
||
|
Figure 4 BETA South Afrika variant
Genome and Spike Fibonacci UA/CG metastructures |
||
|
|
|
|
Figure 6 DELTA India B1.617.2 variant
Genome and Spike Fibonacci UA/CG metastructures |
Seven
OMICRON strains
|
|
|
Figure 7 OMICRONSA1 variant from South
Afrika Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 8 OMICRONSA2 variant from South
Afrika Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 9 OMICRONSA3 variant from South
Afrika Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 10 OMICRONCAL first case variant from USA (California)
Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 11 OMICRONMIN second case variant
from USA (Minesota) Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 12 OMICRONCAN first case variant
from CANADA Genome and Spike Fibonacci UA/CG metastructures |
|
|
|
Figure 13 OMICRONBEL first case variant
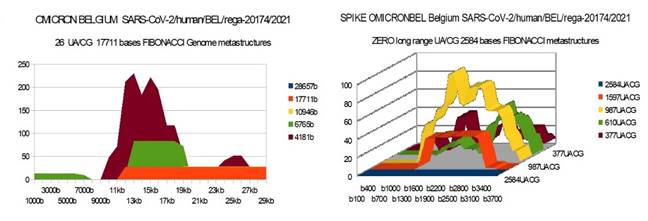
from EUROPA (Belgium) Genome and Spike Fibonacci UA/CG metastructures |
The analysis of the graphs of these 13 virus strains leads to 3 major conclusions that we will classify here according to their order of possible decreasing epidemiological significance:
1) At the level of the Spike, ALL the longest Fibonacci meta-structures covering 2/3 of the Spike sequence (2584 AU / CG, in blue) completely disappear for ALL the OMICRON cases analyzed (Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13). This had never happened for any of the previous variants (including bat RATG13 and Wuhan spikes).
This property (the long-range meta-structures) must reinforce the structure stability of the spike's RNA, and therefore possibly contributes to increased INFECTIOSITY via efficient replication.? Then, according to this principle, all OMICRON strains should have a lower INFECTIOSITY than other initial variants, DELTA included.?
2) In a previous work Perez (2021a), we noticed the typical "podium" structure of the 1597 AU / CG metastructures (red). We had suggested the fact that this podium form could result from a "hairpin" conformation of the spike RNA, a property likely to reinforce its structure and lifespan, therefore its INFECTIOSITY? Let's compare the presence of these podiums in Figure 3, Figure 4, Figure 5, Figure 6 with their total absence in OMICRON in Figure 7, Figure 8, Figure 9, Figure 10, Figure 11, Figure 12, Figure 13.
Hence, the hairpin structure may be abrogated for the OMICRON spikes (see images). The RNA of the OMICRON spikes would be less stable and therefore LESS INFECTIOUS than for the previous variants.
3) The very long meta-structures of 17,711 bases covering 2/3 of the whole genome (in red on the genome graphs located to the left of the images), though they are high for the previous variants, here become very diverse for the OMICRON genomes. We may have to look for the causes, in particular there are only 4 meta-structures of 17,711 bases for 2 cases of American OMICRON. What would this mean in the case of a very little infectious and pathogenic virus?
|
Table 2 Synthesis of cross-homologies and differences between the 7 OMICRON Spike sequences |
||||||
|
SPIKES
Cross homologies |
SA2 |
SA3 |
CAL |
MIN |
CAN |
BEL |
|
(Number of
different nucleotides) |
||||||
|
SA1 |
0 |
1 |
2442 |
1889 |
0 |
0 |
|
SA2 |
1 |
2442 |
1889 |
0 |
0 |
|
|
SA3 |
2443 |
1890 |
1 |
1 |
||
|
CALifornia |
2481 |
2442 |
2442 |
|||
|
MINesota |
1889 |
1889 |
||||
|
CANada |
0 |
|||||
|
Notes: The differences for 1889 and 2442
come from local deletions and shifts |
||||||
We can therefore see that the Canada and Belgium OMICRON spikes are identical. It is the same between SA1 and SA2 which are identical and have only one mutation compared to SA3.
On the other hand, one could think that the insert of 3 amino acids "EPE" in the Spike protein could have influenced the remarkable differentiation of OMICRON with respect to the other variants.
We tested the scenario of a spike without the EPE insert. In particular, the absence of these 3 amino acids could restore the very long 2584 AU / CG meta-structures of the Spike.
Also, we confirm that the genome and spike profiles generated in that scenario retainother characteristics of the graphs presented here.
The emergence of “21L”, a second sister-clade
lineage of the OMICRON variant
At the beginning of December 2021, a second sister-clade of the variant appeared, called 21L, that turns out to be very different from the original 21K branch that initially appeared in South Africa.
The differences between these 2 branches can be found in https://covariants.org/variants/21K.Omicron
All the other results presented here concerned the original 21K branch.
We however analyzed 8 additional 21L genomes obtained by Blast with the Prion region as a primer.
USA1 Sequence ID: OL822696.1 USA/MA
USA2 Sequence ID: OL819480.1 USA/MA
SW Sequence ID: OV145235.1 Switzerland/BL
USA4 Sequence ID: OL717063.1 USA/CA
USA5 Sequence ID: OL819774.1 USA/RI
USA6 Sequence ID: OL815452.1 USA/CA
USA7 Sequence ID: OL800690.1 USA/PA
UK Sequence ID: OV111076.1 UK
In our opinion, despite a very large divergence between 21K and 21L (mutations and Fibonacci metastructures) the important point concerns the persistence of mutations in the Prion-like region that remains COMMON to the 2 branches 21K and 21L.
In this new branch 21L we will note in particular that these 4 deletions & mutations S: L24-, S: P25-, S: P26-, and S: A27S are exactly localized in the 225 bases region at the very beginning of the Spike gene and where we identified 4 small HIV EIE fragments (such as in HIV1 KENYA) Perez and Montagnier (2020).
Table 3 contains 2 South African 21K lines and 6 new 21L lines. It shows the major differences between the 2 sub-lineages 21K and 21L. At the genome level, the values remain very heterogeneous as in 21K. At the level of Spikes, unlike 21K (South Africa) the 21L line contains long metastructures of 2584 AU / CG. On the other hand, the "Podiums" of the 1597 AU / CG disappear in 21L as it is already the case in 21K South Africa.
|
Table 3 Second OMICRON 21L lineage - summary table of the number of longest UA / CG FIBONACCI metastructures of Genomes (17711 bases) and Spikes (2584 bases) |
||||
|
Reference |
Long range Genomic 17711 UA/CG metastructures |
Long range Spike 2584 UA/CG metastructures |
Long range Spike « Podium like » 1587 UA/CG metastructures |
Notes |
|
Bat RaTG13 |
26 |
40 |
28 59 31 (a) |
(a) ALL « podium like » 1597 UA/CG structures |
|
Initial Wuhan first sequence |
8 |
7 |
37 64 26 |
|
|
Wuhan Lineage D614G |
8 |
5 |
18 44 26 |
|
|
ALPHA UK variant |
28 |
12 |
25 51 26 |
|
|
BETA South Afrika |
35 |
12 |
25 51 26 |
|
|
GAMMABrazil |
48 |
10 |
19 34 15 |
|
|
CALifornia variant CAL20C |
5 |
6 |
44 70 26 |
|
|
MINK |
5 |
6 |
41 67 26 |
|
|
MARSEILLE4 |
33 |
5 |
18 44 26 |
|
|
B1.617 INDIA |
53 |
12 |
41 67 26 |
|
|
B.1.617.2 DELTA |
34 |
7 |
30 54 24 |
|
|
OMICRON 21L lineage |
||||
|
26 |
7 |
43 |
21L |
|
|
48 |
7 |
45 |
21L |
|
|
6 |
32 |
91 |
21L |
|
|
34 |
0 |
36 |
21K |
|
|
28 |
7 |
45 |
21L |
|
|
54 |
0 |
36 |
21K |
|
|
0 |
14 |
46 |
21L |
|
|
20 |
20 Nota |
48 |
21L |
|
|
Nota: Between the MINESOTA OMICRON (Table 1 and Figure 11) and the UK
strain from Table 3, there is only 1 UCAG nucleotide difference in Spike
sequence which provides a variation of 5 long range 2584 UA/CG metastructures
(20 for UK instead 15 for Minesota). |
||||
4. DISCUSSION
Genome instabilities and epidemic endings are a
general law of virus evolution
The mechanisms by which the genome of a rapidly mutating virus may become unstable and provoke the end of an epidemic are unknown, but it seems that they do exist in parallel with the development of the collective immunity or as a consequence of it. We are referring here to viruses that mutate at a rate much faster than the human life span, like flu viruses and SARS coronavirus, excluding retroviruses that can mutate extremely rapidly like the HIV virus because they integrate the genome and thus infect permanently their host. The collective immunity tends to exert a positive evolutionary pressure on these classical RNA viruses to force their adaptation to the more resistant fraction of the population apparently untouched by the virus, despite having been in contact with it, or to the population already immunized after having developed marked symptoms. Ter Meulen (2006)[ter Meulen, Marco Vignuzzi]
However, it is observed until now that the efficiency of a perpetual adaptation process is not guaranteed, fortunately, and as a matter of fact the acute phase of epidemics always fades away even-though virus variants continue to circulate for quite a number of years, having a marginal lethal impact. This was the case with the Spanish Influenza A deadly world pandemic of 1918-1919, the basis of the pathogenicity of which remain unanswered and that disappeared progressively. Research indicates that descendants of the 1918 virus still persists enzootically in pigs and probably also circulated continuously in humans, undergoing gradual antigenic drift and causing annual epidemics, until the 1950s. Taubenberger (2006)[Taubenberger]
This fact observed for the Spanish Influenza H1N1 virus has a direct and profound implication on the understanding of the stability of viral genome. Would the immunity escape process be always guaranteed then the virus would circulate endlessly along the years with potentially the same level of contagiousness and pathogenic character? The fact that does not seem to happen means that viruses that mutate much faster than the human lifespan do not possess a capacity of infinite adaptation. As the collective immunity spreads, they cannot continuously generate new viable and efficient variants issued from mutation and recombination of themselves and end up generating defective genomes Bosma (2019), Rezelj (2021), Nayak (1989). To survive this programmed decline, they need to re-assort with a viable “helper” virus, a situation not necessarily fulfilled. Vignuzzi (2019)[Marco Vignuzzi, Von Magnus]
Thus, it is logical to hypothesized that having explored all possible mutations the genomes of SARS-CoV2 will inevitably become defective and not able anymore to generate new efficient variants in the absence of such a rescue mechanism. It may thus become dormant in an intermediate host until it disappears and/or be replaced by another virus. There is a priori no scientific reason to think otherwise despite the fact that individual lock-down and mass vaccination have biased the evolutionary pressure on the virus giving it more time to find adaptive mutation/re-assortment. It must consider that the deadly epidemic of SARS-CoV of 2003-2004, by many aspects a virus close to SARS-CoV2, has ended without the need for mass vaccination and lock-down. The end of the epidemic was marked by an increasingly defective genome with progressive deletion in the accessory Orf8 gene, at the end of the genome sequence, that participates in viral replication. “A 29 nucleotide (nt) deletion within ORF8 occurred in all strains involved in the middle and late phase of the human epidemic” Muth (2018), Chinese SARS Molecular Epidemiology Consortium (2004)[Doreen Muth, Rossa, Chinese SARS Molecular Epidemiology Consortium]. Truncated genomes at the level of the terminal Orf coding the multifunctional-role nucleocapside protein N have also been observed at the end of the epidemic. Muth (2018), McBride (2014)[Ruth McBride] The hampered virions did not have replicative capabilities.
The reason why deletion and truncation may occur at the 3' extremity of the viral genome is not known. One theoretical hypothesis is that the viral replicase generates many copy errors or arrests in this end section due to an overall destabilization of the RNA strand. This may be caused by the disruption of the cohesive electrostatic interactions at medium and long-range with dynamical implication for the coherence of the RNA structure, all that being forced by the evolutionary pressure mutations needed to escape immune resistance. Perez (1991), Chen (2016) The mechanism certainly involves destabilization of the mRNA secondary structure, involving complementary base pairing of the RNA single strand folding on itself to form "hairpin" conformations, probably making it more fragile.
From DELTA to OMICRON, variants exhibit an increased
number of mutations in the spike protein gene
The delta variant of SARS-CoV-2, B.1.617.2, has 23 mutations compared to the first identified COVID-19 strain (alpha strain) Hodcroft (2021). Twelve of those mutations are in the spike protein. One study has reported that the delta variant is 60% more transmissible than the alpha variant. As of August 2021, the delta variant has quickly become the dominant strain Hodcroft (2021), Shiehzadegan (2021) [Hodcroft, Shiehzadegan].
The number of non-synonymous mutations found in the spike of the Omicron variant is exceeding by far that found in other variants of concern. “Non-synonymous mutations were identified in the spike (S)‒encoding (n = 35) and other viral protein‒encoding (n = 22) regions. Among the nonsynonymous mutations in the S protein, 43% (n = 15) were also identified in other VOCs/variants of interest, and 31% (n = 11) were found only in VOCs (Alpha, n = 6; Beta, n = 4; Gamma, n = 5; Delta, n = 4). Some of the point mutations and deletions found in other regions are not novel and can also be found in other variants at different frequencies” Gu (2021).
The
main question of the PRION-Like region pending risk in the OMICRON Spike
Remarkably, we note 8 mutations (see figure below) in the prion-like region of the spike gene. This is a remarkable observation because this region that represents less than 3% of the spike genome gathers a fourth (25%) of all the non-synonymous mutations. What does that mean? In fact, the prion-region of the spike plays an essential role in cell adhesion and entry and therefore possibly as well in the blood-brain barrier crossing Tetz and Tetz (2020), Buzhdygan (2020), Reynolds (2021)[Tetz, Buzhdygan, Reynolds]. This concentration of mutation confirms this gene region is a key element in the infectiousness of the virus and that the virus is subjected to a strong adaptation pressure.
We looked for the possible presence of this Prion region and then possibly of these 8 mutations characterizing OMICRON in the spikes of different strains of sequenced genomes of bat SARS-like coronaviruses and SARS-CoV2 strains and variants.
Let us recall here that this small Prions region measures only 114 bases, i.e.,38 amino acids. First, we find that this region is completely absent from the 2 strains covzx45 and covzxc21.
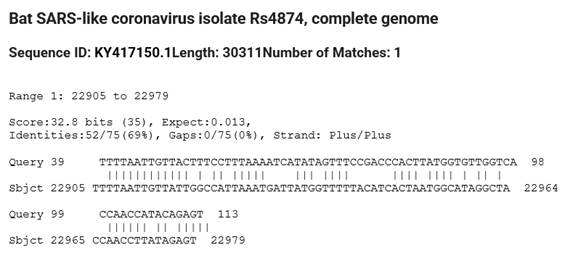
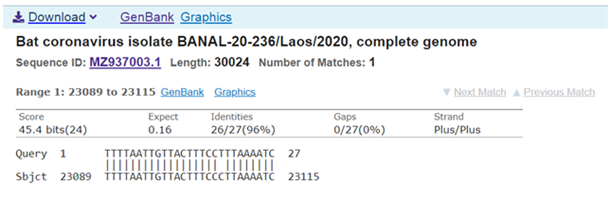
On the other hand, a Blast search in bat SARS-Like coronaviruses with the OMICRON Prion region as a search criterion only identified a single strain: Rs4874. The homology is however reduced to the Prion region beyond base 39 on the 114 base-long Prion-Like region (Table 4, Table 5).
|
Table 4 Partial 75
bases homology of Bat coronavirus Rs4874 isolate with the 114 bases Prion
region |
|
|
|
Table 5 Other
partial 27 bases homology of Bat coronavirus from LAOS isolate with the 114
bases Prion region |
|
|
A third strong homology was found with Bat RaTG13 Spike sequence:
Between both Prion regions of OMICRON and BatRaTG13; there are 81 common nucleotides (on 114) with 71% identity.
Recall




The 8 amino acids mutations are underlined: 6 are different at the nucleotides level. 6 also are different at the amino acids level.
Finally, a summary analysis of the Prion region in variants ALPHA, BETA, GAMMA and DELTA provides in all cases the same difference of 7 amino acids with respect to the 7 studied OMICRON and of 8 amino acids with respect to the reference OMICRON.
|
|
|
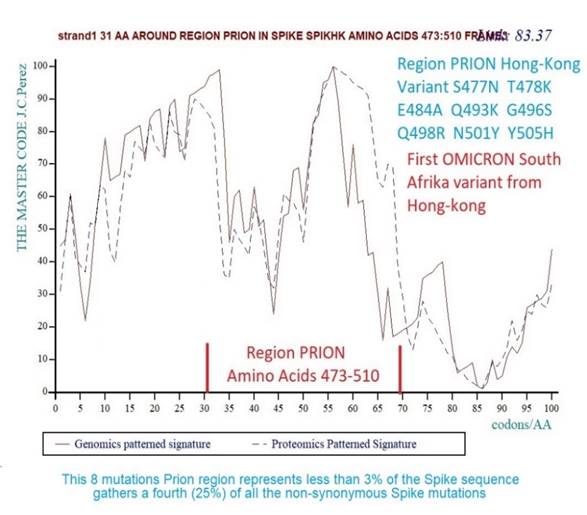
Figure 14 Region PRION in the OMICRON
Spike with its 8 amino acids mutations. This is a remarkable observation
because this region that represents less than 3% of the spike genome gathers
a fourth (25%) of all the Spike non-synonymous mutations |
Figure 14 above shows the Genomics / Proteomics "Master code" coupling Perez and Montagnier (2021) in the Prion region of Spike for the Omicron sequence of the first South Afrikan variant discovered in a patient from Hong Kong.
A rapid analysis using the PLAAC Prions region search software (http://plaac.wi.mit.edu) confirms the presence of this Prions region reported by (Tetz 2020) in the respective Spike of SARS-CoV2 Wuhan, the successive variants including DELTA as well as in the 2 mRNA vaccines Moderna and Pfizer. On the other hand, this Prion regio n TOTALLY disappears from the new OMICRON 21K (SA1) and 21L (Minesota) variants.
Loss of Fibonacci long-range fractal meta-structures coherence in OMICRON variants
|
|
|
Figure 15 The 2 longest Fibonacci Spike
UA/CG Metastructures are either suppressed (2584 UA/CG) or reduced (1594
US/CG « PODIUMS » top value) in all 7 OMICRONS variants analyzed with the
exception of MINESOTA OMICRON Strain which is a different sub-clade. |
It is mathematically demonstrated that a hidden fractal order exists in DNA and RNA genomic sequences Perez (1991), Perez (2018), Perez (2021a), Perez (2021b) . It consists of long-range Fibonacci meta-structures that are being thought to be associated with genome overall stability in relation with their conformational structure and dynamic.
When we apply the Fibonacci meta-structure analysis to OMICRON, we observe the presence of theses Fibonacci metastructures (except California case) at the level of the whole genome remain high, indicating a variant probably contagious with a level analog to DELTA). Effectively these metastructures traduce genome adaptation to the human host, they have increased with successive variants from the Wuhan initial strain.
Meanwhile the 7 spikes are ALL very weak in terms of long-range meta-structures compared to the Wuhan spike and ALL the other variants.
Would this result in very low pathogenic character as the first observations seem to indicate in South Africa?
Due the variable extent and timing of mass vaccination across countries along the year 2021 it is impossible to decipher whether the increased number of mutations along with enhanced contagiousness has actually corresponded to a decreased pathogenic character of the variant Delta. Countries like Israel and USA that had completed a level of full vaccination (2 doses) in 50% of the population by the end of March and July, respectively. However, these 2 countries have both experienced dramatic waves of Covid-19 related deaths with the Delta variant beginning at the end of July Shiehzadegan (2021)[Shiehzadegan]. Whereas, in France, at the end of July as vaccination had reached hardly 48% of 2-doses, after having been lagging behind with only 4% 2d vaccination at the end of March, the virus death toll was extremely weak compared with Israel and USA. This was the case as well in many other European countries and seems to indicate a lower pathogenic character. Proponents of the catastrophic epidemic scenario would argue that this low death toll is the result of the enforcement of the use vaccination and of the sanitary pass in France and European countries.
With the third vaccine injection becoming mandatory by January 15, 2022, for everybody over 18 years in France and many European countries the same scenario of denial of the possible natural ending of the epidemic may arise together with an impossibility to measure the real level of pathogenic character of this mutant. It may however be possible to measure it in the coming months in some USA states another country in the world where a third injection is not going to be mandatory.
Regarding the question of the possible contagiousness, infectiousness and pathogenic character of OMICRON, we compared our results of Fibonacci metastructures at the scale of whole genomes with this very recent article by Cosic et al. (2021).
Of note, in contrast to our approach, based on the analysis of viral genomes, other innovating physical models such as the Resonant Recognition Model (RRM) are attempting to evidence difference in contagiousness and infectivity of variants directly studying the spike protein interaction with the ACE2 receptor. The RRM indicates a lesser infectiousness of Omicron vs Delta.
The main difference between our results and those described above come from the 2 American strains California and Minesota of OMICRON which, in our opinion, differ radically from the 5 others studied with very weak Fibonacci metastructures at the genome scale (only 4 UA/CG meta structures of 17,711 bases in both cases of California and Minesota OMICRON variants).
On the other hand, it will be noted that the Prion region of the Spike also plays, by its position, a major role in the infectiousness process.
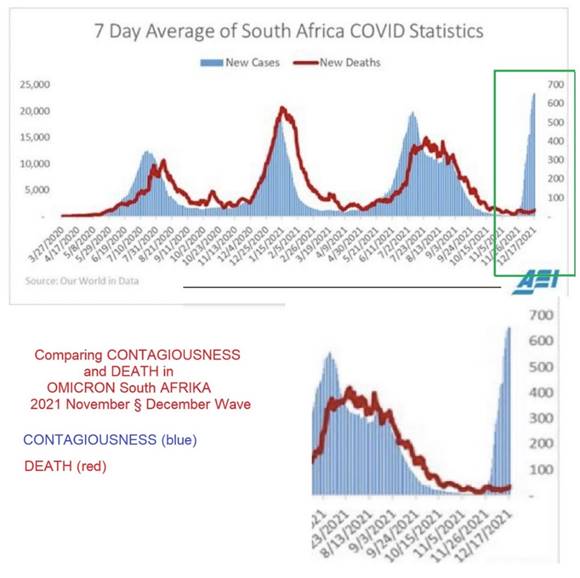
The first observations show us that OMICRON is very contagious although probably less infectious than previous variants (Figure 16).
The fact that this Prion region has mutated a lot in OMICRON but also the fact that the Prion effect is based on a fine-tuning between the relative proportions of the 2 amino acids Q and N constitutes a key element.
The high infectiousness of SARS-CoV2 before variant Delta could therefore be based on this region since at the level of Fibonaccis metastructures the Spike is now very destructured (original 21K line) although it begins to restructure in the last 21L sub-lineage, a sign of a re-adaptation to its human host with a possible relative regain of infectiousness.
|
|
|
Figure 16 Comparing CONTAGIOUSNESS and
DEATH in OMICRON South Afrika 2021 November § December Wave; CONTAGIOUSNESS
(blue) and DEATH (red) |
5. CONCLUSIONS
Contrary to the emergence of the original SARS-CoV2 virus in Wuhan in December 2019 that poses serious concerns regarding its natural origin, Perez and Montagnier (2020), Lounnas (2021), Quay (2020)Lounnas, Quay], we are here witnessing a scenario that is fully in accordance with established knowledge on epidemic evolution.
The number of mutations in the spike gene increasing dramatically along with contagiousness from the Alpha to the Omicron variant is in clear agreement what we know on natural pandemic development such as that of the Spanish influenza of 1918-1919. End of epidemic variants are expected to be less infectious and highly defective.
It is not impossible that Omicron is a end of epidemic variant that has tried to survive by recombining with a helper virus (a benin coronavirus), possibly via HIV infected persons who are more prone to develop long-term viral infection but also who may integrate the vaccine RNA in their genome via the HIV reverse transcriptase.
And of course, end of epidemic variants normally is much less pathogenic but highly contagious helping the collective immunity to spread broadly. This article in Nature explains how debilitated variants may serve as natural vaccine Rezelj (2021).
This should be reflected in a worldwide continuously decreasing Covid-19 death toll in the coming months, a decrease already primed with the variant delta, despite the fact that Pifzer has already admittingly announced its vaccines will provide less immunity against it than other variants Sguazzin (2021)[Sguazzin].
6. ACKNOWLEDGEMENTS
Thanks for sending us the three first OMICRON genomes sequences from South Afrika:
The three original South Afrika sequences received from the team Pr Tulio de Oliveira, Dr Penny Moore and Dr Cathrine Scheepers (HIV & SARS-CoV-2 Virology Section, Centre for HIV & STI’s, National Institute for Communicable Diseases (NICD), a Division of the NHLS SAMRC Antibody Immunity Research Unit (AIRU), 1 Modderfontein Road, Sandringham, 2131, South Africa).
Thanks for sending us the first OMICRON genome sequence in USA from California:
The first California sequence received from Professor Charles Chiu, M.D./Ph.D. Professor, Laboratory Medicine and Medicine / Infectious Diseases Director, UCSF-Abbott Viral Diagnostics and Discovery Center Associate Director, UCSF Clinical Microbiology Laboratory UCSF School of Medicine.
Thanks for fructuous discussions about this article to Megawaty Tan (A private researcher based in South Sumatera, Indonesia).
We thank particularly dr Richard M Fleming PhD, MD, JD (https://www.flemingmethod.com/ and https://www.francesoir.fr/amp/article/videos-les-debriefings/dr-richard-fleming-son-debriefing) for discussions on SARS-CoV2 origins and prion like diseases risk (see https://biomedres.us/fulltexts/BJSTR.MS.ID.000369.php).
Finally, this work is the result of multiple exchanges and advice, since the very beginning of the COVID-19 pandemic, for which we must thank Professor Luc Montagnier (Nobel prizewinner for his discovery of HIV, Fondation Luc Montagnier Quai Gustave-Ador 62 1207 Geneva, Switzerland).
REFERENCES
Cosic et al (2021) Irena Cosic et al, Alnalysis of Omicron SARS-CoV-2 Variant Using Resonant Recognition Model ; December 2021, DOI: 10.13140/RG.2.2.17453.00484. Retrieved from https://www.researchgate.net/publication/356655056_Analysis_of_Omicron_SARS-CoV-2_Variant_Using_Resonant_Recognition_Model
Bosma (2019) Trent J. Bosma, Konstantinos Karagiannis , Luis Santana-Quintero et al. Identification and quantification of defective virus genomes in high throughput sequencing data using DVG-profiler, a novel post-sequence alignment processing algorithm Plos One Published: May 17, 2019. Retrieved from https://doi.org/10.1371/journal.pone.0216944
Buzhdygan (2020) Tetyana P. Buzhdygan, Brandon J. DeOre Abigail, Baldwin-Leclair et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier Neurobiology of Disease 146 (2020) 105131.
Chen (2016) Chuanying Chen and B. Montgomery Pettitt, DNA Shape versus Sequence Variations in the Protein Binding Process Biophys J. 2016 Feb 2; 110(3): 534–544.Published online 2016 Feb 2. doi: 10.1016/j.bpj.2015.11.3527
Chinese SARS Molecular Epidemiology Consortium (2004) Chinese SARS Molecular Epidemiology Consortium Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004). DOI:10.1126/science.1092002
Chiu (2005) Rossa W.K. Chiu,Stephen S.C. Chim,Yu-kwan Tong et al. Tracing SARS-coronavirusvariant with large genomic deletion. Emerging infectious diseases 11, 168–170 (2005). doi: 10.3201/eid1101.040544 . Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3294368 /
Gu (2021) Haogao Gu, Pavithra Krishnan, Daisy Y.M. Ng, et al. Probable Transmission of SARS-CoV-2 Omicron Variant in Quarantine Hotel, Hong Kong, China, EID Journal November 2021 Volume 28, Number 2—February 2022 . Retrieved from https://wwwnc.cdc.gov/eid/article/28/2/21-2422_article
Hodcroft (2021) Hodcroft, E.B. SARS-CoV-2 Mutations and Variants of Interest. CoVariants. Available online: https://covariants.org/ (accessed on 9 October 2021).
Lapid (2021) Nancy Lapid Reuters Omicron variant may have picked up a piece of common-cold. Retrieved from https://www.reuters.com/business/healthcare-pharmaceuticals/omicron-variant-may-have-picked-up-piece-common-cold-virus-2021-12-03/
Los Angeles Time (2021) Los Angeles Time Did failure to adequately treat HIV patients give rise to the Omicron variant? Retrieved from https://www.latimes.com/science/story/2021-12-02/did-omicron-coronavirus-variant-arise-in-patient-with-uncontrolled-hiv
Lounnas (2021) Valère Lounnas Li-Meng Yan in the spotlights with embarrassing questions to some world-renowned scientists and to expert virologists of the Pasteur Institute Zenodo. Retrieved from https://zenodo.org/record/4664546#.YbCuxVXMLBo
Lounnas (2021) Valère Lounnas et Gérard Guillaume Histoire du COVID-19 – chapitre 9, partie 2 : Où le mathématicien Jean-Claude Perez et le professeur Luc Montagnier nous révèlent que les laboratoires de virologie font planer un danger obscur sur l'humanité. Retrieved from https://www.francesoir.fr/histoire-du-covid-19-chapitre-9-partie-2
Lounnas (2021) Valère Lounnas, Gérard Guilllaume et Xavier Azalbert Histoire du Covid-19, la pandémie de toutes les peurs Amazon -Kindle
Matalon (2021) Vincent Matalon Variant Omicron : la course contre la montre des chercheurs pour percer les secrets de cette mutation "préoccupante" du coronavirus. Retrieved from https://www.francetvinfo.fr/sante/maladie/coronavirus/variant-omicron/variant-omicron-la-course-contre-la-montre-des-chercheurs-pour-percer-les-secrets-de-cette-mutation-preoccupante-du-virus_4865785.html
McBride (2014) Ruth McBride, Marjorie van Zyl,and Burtram C. Fielding The Coronavirus Nucleocapsid Is a Multifunctional Protein Viruses. 2014 Aug; 6(8): 2991–3018. Published online 2014 Aug 7. doi: 10.3390/v6082991
Megawaty Tan et al, (2021) - Megawaty Tan, Valère Lounnas, Xavier Azalbert, Christian Perronne. May vaccines select SARS-CoV-2 variants more readily escaping immunity - an analysis of public data. Archives of Microbiology & Immunology 5 (2021): 316-324. Retrieved from https://www.fortunejournals.com/articles/may-vaccines-select-sarscov2-variants-more-readily-escaping-immunity.pdf
Muth (2018) Doreen Muth, Victor Max Corman, Hanna Roth, Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission Published:11 October 2018 Open Access. Retrieved from https://www.nature.com/articles/s41598-018-33487-8
Nayak (1989) Debi P. Nayak, Thomas M. Chambers and Ramesh K. Akkina Structure of Defective-Interfering RNAs of Influenza Viruses and Their Role in Interference The Influenza Viruses pp 269-317. Retrieved from https://link.springer.com/chapter/10.1007/978-1-4613-0811-9_6
Perez (1991) J.C. Perez (1991), "Chaos DNA and Neuro-computers: A Golden Link", in Speculations in Science and Technologyvol. 14 no. 4, ISSN 0155-7785, January 1991Speculations in Science and Cell Motility 14(4):155-7785, Retrieved from https://www.researchgate.net/publication/258439719_JC_Perez_1991_Chaos_DNA_and_Neuro-computers_A_Golden_Link_in_Speculations_in_Science_and_Technologyvol_14_no_4_ISSN_0155-7785
Perez (2010) Perez JC. Codon populations in single-stranded whole human genome DNA Are fractal and fine-tuned by the Golden Ratio 1.618. Interdiscip Sci. 2010 Sep;2(3):228-40. doi: 10.1007/s12539-010-0022-0. Epub 2010 Jul 25. Erratum in: Interdiscip Sci. 2010 Dec;2(4):373. PMID: 20658335.
Perez (2018a) Perez JC., Neuroblastoma and Glioblastoma Brain Cancers: «Human Genome Optimum» (HGO) a Global Genome Strategy controlling all Human Chromosome LOH Deletions, Published: March 19, 2018, DOI: 10.31031/NACS.2018.01.000512
Perez (2018b) Jean-Claude Perez. Towards a Universal Law Controlling all Human Cancer Chromosome LOH Deletions, Perspectives in
Perez (2018c) Prostate and Breast Cancers screening. Canc Therapy & Oncol Int J. 2018; 9(2): 555756. DOI: 10.19080/CTOIJ.2018.09.555756
Perez (2018d) - Perez JC (2018) Sapiens Mitochondrial DNA Genome Circular Long Range Numerical Meta Structures are Highly Correlated with Cancers and Genetic Diseases mtDNA Mutations. J Cancer Sci Ther 9: 512-527. doi: 10.4172/1948- 5956.1000469. Retrieved from https://www.hilarispublisher.com/open-access/sapiens-mitochondrial-dna-genome-circular-long-range-numericalmeta-structures-are-highly-correlated-with-cancers-and-genetic-disea-1948-5956-1000469.pdf
Perez (2020) - Jean-Claude PEREZ. (2020). WUHAN COVID-19 SYNTHETIC ORIGINS AND EVOLUTION. International Journal of Research - Granthaalayah, 8(2), 285–324. Retrieved from https://doi.org/10.5281/zenodo.3724003
Perez and Montagnier (2020) Perez, J. C. , Montagnier, L., COVID-19, SARS AND BATS CORONAVIRUSES GENOMES PECULIAR HOMOLOGOUS RNA SEQUENCES , International Journal of Research -GRANTHAALAYAH: Vol. 8 No. 7 (2020): Volume 8 Issue 7: July 2020
Perez (2021a) Jean-Claude Perez, SARS-COV2 VARIANTS AND VACCINES MRNA SPIKES FIBONACCI NUMERICAL UA/CG METASTRUCTURES , International Journal of Research -GRANTHAALAYAH: Vol. 9 No. 6 (2021): Volume 9 Issue 6: June 2021
Perez (2021b) - Perez, J. C. (2021). THE INDIA MUTATIONS AND B.1.617 DELTA VARIANTS: IS THERE A GLOBAL "STRATEGY" FOR MUTATIONS AND EVOLUTION OF VARIANTS OF THE SARS-COV2 GENOME?. International Journal of Research -GRANTHAALAYAH, 9(6), 418–459. Retrieved from https://doi.org/10.29121/granthaalayah.v9.i6.2021.4039
Perez and Montagnier (2021) - Perez, J. C., & Montagnier, L. . (2021). SIX FRACTAL CODES OF LIFE FROM BIOATOMS ATOMIC MASS TO CHROMOSOMES NUMERICAL STANDING WAVES: THREE BREAKTHOUGHS IN ASTROBIOLOGY, CANCERS AND ARTIFICIAL INTELLIGENCE. International Journal of Research -GRANTHAALAYAH, 9(9), 133–191. Retrieved from https://doi.org/10.29121/granthaalayah.v9.i9.2021.4191
Perez et al. (2021) - Jean-Claude Perez, Valère Lounnas, Megawaty Tan, Xavier Azalbert, Christian Perronne - "May the SARS-CoV-2 OMICRON variant signal the end of the pandemic – a Fibonacci fractal analysis", to be published in Archives of Microbiology & Immunology.
Quay (2020) Steven Carl Quay, A Bayesian analysis concludes beyond a reasonable doubt that SARS-CoV-2 is not a natural zoonosis but instead is laboratory derived Zenodo. Retrieved from https://zenodo.org/record/4477081#.YbCsolXMLBo
Racaniello (2019) Vincent Racaniello A lesson from SARS-CoV for 2019-nCoV. Retrieved from https://www.virology.ws/2020/01/23/a-lesson-from-sars-cov-for-2019-ncov/
Rezelj (2021) Veronica V. Rezelj, Lucía Carrau, Fernando Merwaiss, et al. Defective viral genomes as therapeutic interfering particles against flavivirus infection in mammalian and mosquito hosts Nature Communications 12, Article number: 2290 (2021). Retrieved from https://www.nature.com/articles/s41467-021-22341-7
Reynolds (2021) Jessica. L. Reynolds and Supriya D. Mahajan SARS-COV2 Alters Blood Brain Barrier Integrity Contributing to Neuro-Inflammation J Neuroimmune Pharmacol. Jan 6 : 1–3 2021 [Epub ahead of print] doi: 10.1007/s11481-020-09975-y
Sguazzin (2021) Antony Sguazzin and Jason Gale Pfizer Shot Provides Partial Omicron Shield in Early Study . Retrieved from https://www.bloomberg.com/news/articles/2021-12-07/omicron-doesn-t-escape-pfizer-vaccine-completely-lab-head-says
Shiehzadegan (2021) Shayan Shiehzadegan, Nazanin Alaghemand, Michael Fox and Vishwanath Venketaraman Analysis of the Delta Variant B.1.617.2 COVID-19 Clin Pract 2021 Oct 21;11(4):778-784. doi: 10.3390/clinpract11040093.
Taubenberger (2006) Jeffery K. Taubenberger and David M. Morens 918 Influenza: the Mother of All Pandemics Emerg Infect Dis. 2006 Jan; 12(1): 15-22. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3291398/
Ter Meulen (2006) ter Meulen J, van den Brink EN, Poon LLM et al. Human monoclonal antibody combination against SARS coronavirus : synergy and coverage of escape mutants, PLoS Med 2006 Jul;3(7):e237; doi: 10.1371/journal.pmed.0030237.
Tetz and Tetz (2020) George Tetz and Victor Tetz SARS-CoV-2 Prion-Like Domains in Spike Proteins Enables Higher Affinity to ACE2 Preprint Research Gate March 2020 DOI:10.20944/preprints202003.0422.v1
Venkatakrishnan (2021) AJ Venkatakrishnan, Praveen Anand, Patrick J Leneha t al. Omicron variant of SARS-CoV2 harbors a unique insertion mutation of putative viral or human genomic origin OFSPreprint . Retrieved from https://osf.io/f7txy/?s=03
Vignuzzi (2019) Marco Vignuzzi, Carolina B López Defective viral genomes are key drivers of the virus-host interaction Review Nat Microbiol. 2019 Jul;4(7):1075-1087. doi: 10.1038/s41564-019-0465-y. Epub 2019 Jun 3.
 This work is licensed under a: Creative Commons Attribution 4.0 International License
This work is licensed under a: Creative Commons Attribution 4.0 International License
© Granthaalayah 2014-2021. All Rights Reserved.