
THE INDIA MUTATIONS AND B.1.617 DELTA VARIANTS: IS THERE A GLOBAL "STRATEGY" FOR MUTATIONS AND EVOLUTION OF VARIANTS OF THE SARS-COV2 GENOME?Jean-Claude Perez 1*
|
|
|
|
Article Type: Research Article
Article Citation: Jean-Claude
Perez. (2021). THE INDIA MUTATIONS AND B.1.617 DELTA VARIANTS: IS THERE A
GLOBAL "STRATEGY" FOR MUTATIONS AND EVOLUTION OF VARIANTS OF THE
SARS-COV2 GENOME? International Journal of Research -GRANTHAALAYAH, 9(6), 418-459.
https://doi.org/10.29121/granthaalayah.v9.i6.2021.4039
Received Date: 15 June 2021
Accepted Date: 30 June 2021
Keywords:
India
Global
Strategy
Evolution
Genome
ABSTRACT
In this paper, we
run for all INDIA mutations and variants a biomathematical numerical method for
analysing mRNA nucleotides sequences based on UA/CG
Fibonacci numbers proportions (Perez, 2021). In this
study, we limit ourselves to the analysis of whole genomes, all coming from the
mutations and variants of SARS-CoV2 sequenced in India in 2020 and 2021. We
then demonstrate - both on actual genomes of patients and on variants combining
the most frequent mutations to the SARS-CoV2 Wuhan genomes and then to the
B.1.617 variant - that the numerical Fibonacci AU / CG metastructures
increase considerably in all cases analyzed in ratios of up to 8 times. We can
affirm that this property contributes to a greater stability and lifespan of
messenger RNAs, therefore, possibly also to a greater INFECTUOSITY of these
variant genomes.
Out of a total of
108 genomes analyzed:
·
None
("NONE") of them contained a number of metastructures LOWER than those of the reference SARS-CoV2
Wuhan genome.
·
Eleven
(11) among them contained the same number of metastructures
as the reference genome.
·
97 of
them contained a GREATER number of metastructures
than the reference genome, ie
89.81% of cases. The average increase in the number of metastructures
for the 97 cases studied is 4.35 times the number of SARS-CoV2 UA/CG 17711
Fibonacci metastructures.
Finally, we put a
focus on B.1.617.2 crucial exponential growth Indian variant.
Then, we
demonstrate, by analyzing the main worldwide 19 variants, both at the level of
spikes and of whole genomes, how and why these UA / CG metastuctures
increase overall in the variants compared to the 2 reference strains SARS-CoV2
Wuhan and D614G. Then, we discuss the possible risk of ADE for vaccinated
people.
To complete this
article, an ADDENDUM by Nobelprizewinner Luc
Montagnier was added at the end of this paper.
After various papers related SARS-CoV2 origins and evolution (Perez, 2020) and (Perez§Montagnier, 2020),
in (Perez, 2021), we presented a biomathematical method based on mRNA genomes and spikes UA/CG Fibonacci nucleotides proportions. Particularly we demonstrated a real corelation between variants evolution (UK, South Africa, California, Brazil) and the amount of long-range Fibonacci metastructures.
In order to test this hypothesis, we are interested in the 2 countries in which the effect of variants seems uncontrollable: Brazil and India.
We chose India because the sequencing of genomes is more systematic and reliable there than in Brazil.
For this we proceed in 2 steps:
· Analyzing the first variants of 2020. For this we rely on this publication:
(Muttineri et al, 2021),
https://www.google.com/url?sa=t&source=web&rct=j&url=https://journals.plos.org/plospathogens/article/file%3Fid%3D10.1371/journal.pone.0246173%26type%3Dprintable&ved=2ahUKEwj3zdnZnorwAhUQKBoKHUxnD_EQFjABegQICBAC&usg=AOvVaw1A79ux6UbetoPoRx_jT-Mk
2/ Then we study the most recent changes of 2021. For that we rely on this sydtematic approach:
(Srivastava Surabhi et al, 2021), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7895735/
And more particularly on this Indian GEAR19 database:
https://data.ccmb.res.in/gear19/variants
Consider the sequence of Fibonacci numbers
0 1 1 2 3 5 8 13 21 34 55 89 144 233 377 610 987 1597 2584 4181 6765 10946 17711
28657 46368 75025 121393 196418 317811 514229 832040 1346269 2178309
3524578 5702887...
Example of the SPIKE from Wuhan reference genome, this mRNA SPIKE is 3822 bases UCAG in length.
Recall Wuhan reference https://www.ncbi.nlm.nih.gov/nuccore/NC_045512
Severe acute respiratory syndrome
coronavirus 2 isolate Wuhan-Hu-1, complete genome NCBI Reference
Sequence: NC_045512.2
the longest Fibonacci structures would therefore measure 2584 bases.
When looking for such structures, the first one found is in 1200 location:
therefore, the bases located between 1201 and 3784 (1200 + 2584):
These 2584 bases are broken down respectively into:
1597 bases UA
et 987 bases CG
Here are the first 20 basics that the reader can easily check:

The SPIKE analyzes of this Wuhan-Hu-1 reference genome reports 63 metastructures of this type if we close the sequence on itself (as in mtDNA or bacteria) and 7 metastructures and if we consider the mRNA sequence in its linear form, as will be the case throughout this study.
2.2.1. ANALYZING THE FIRST VARIANTS OF 2020
· Analyzing the first variants of 2020. For this we rely on this publication:
(Muttineri et al, 2021),
https://www.google.com/url?sa=t&source=web&rct=j&url=https://journals.plos.org/plospathogens/article/file%3Fid%3D10.1371/journal.pone.0246173%26type%3Dprintable&ved=2ahUKEwj3zdnZnorwAhUQKBoKHUxnD_EQFjABegQICBAC&usg=AOvVaw1A79ux6UbetoPoRx_jT-Mk
The full-genome viral sequences were deposited in the dataset of GISAID (EPI_ISL_431101, EPI_ISL_431102, EPI_ISL_431103, EPI_ISL_431117, EPI_ISL_438139, EPI_ISL_437626, EPI_ISL_438138) and NCBI GenBank (MT415320, MT415321, MT415322, MT415323, MT477885, MT457402, MT457403).
See also (Govinarajan, 2020a) and (Govinarajan, 2020b).
Now we analyse:
GenBank (MT415320, MT415321, MT415322, MT415323, MT477885, MT457402, MT457403)
Main data source for mutations: https://covariants.org/
2.2.2. ANALYZING 28 INDIAN MUTATIONS APPLIED TO SARS-COV2 WUHAN REFERENCE GENOME
Then we study the most recent changes of 2021. For that we rely on this systematic approach:
(Srivastava Surabhi et al, 2021), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7895735/
And more particularly on this Indian GEAR19 database:
https://data.ccmb.res.in/gear19/variants
We test 2 possible variant scenarios:
If separate mutations are INDIABn,
INDIACn, progressive descent by accumulating mutations by decreasing probabilities.
Example
INDIAC1 = INDIAB1
INDIAC2 = INDIAC1 + INDIAB2
INDIAC3 + INDIAC2 + INDIAB3 ...
…/...
INDIAC28 = INDIAC27 + INDIAB28
Then we study the most recent changes of
2021. For that we rely on this systematic approach:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7895735/
And more particularly on this Indian GEAR19 database:
https://data.ccmb.res.in/gear19/variants
link Table 5%
https://mail.google.com/mail/u/0/#inbox/KtbxLzGLmpFSTVtcKRqRlmnxKrplVzgNnq?projector=1&messagePartId=0.1
2.2.3. ANALYZING 28 INDIAN MUTATIONS APPLIED TO B.1.617 INDIA VARIANT GENOME
We run the same 28 genomes simulations starting from the India variant B.1.617.
2.2.4. SIMULATIONS OF POSSIBLE FUTURE MUTATIONS OF THE VARIANT B.1.617
In (Pragya Yadav et al, 2021), the authors provide a list of the 33 main mutations characterizing the genomes of the Indian variant B 1.617.
On the other hand, we have just studied the impact of the 28 most frequent mutations in India, those which represent more than 5% of contaminations).
It is clear that these 2 sets of mutations partially overlap.
However, it would be interesting to simulate the effect of some of the 28 mutations when they are absent in B.1.617. Indeed, their high frequency makes it possible to suggest their possible future addition to B.1.617.
This is what we will simulate in the last paragraph 3.3.
Now we analyse from (Muttineri et al, 2021):
GenBank (MT415320, MT415321, MT415322, MT415323, MT477885, MT457402, MT457403)
INDIAA1 MT415320
https://www.ncbi.nlm.nih.gov/nuccore/MT415320
INDIAA2 MT415321
https://www.ncbi.nlm.nih.gov/nuccore/MT415321
INDIAA3 MT415322
https://www.ncbi.nlm.nih.gov/nuccore/MT415322
INDIAA4 MT415323
https://www.ncbi.nlm.nih.gov/nuccore/MT415323
INDIAA5 MT457402
https://www.ncbi.nlm.nih.gov/nuccore/MT457402
INDIAA6 MT457403
https://www.ncbi.nlm.nih.gov/nuccore/MT457403
INDIA7 MT477885
https://www.ncbi.nlm.nih.gov/nuccore/MT477885
Table 1: Mutations Table from paper
https://journals.plos.org/plosone/article/figure?id=10.1371/journal.pone.0246173.t003
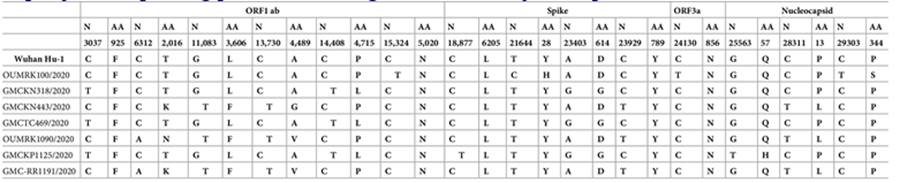
https://journals.plos.org/plosone/article/figure?id=10.1371/journal.pone.0246173.t003
Table 2: Summary Table related 7 SARS-CoV2 real patients from India sequenced in 2020.
|
Reference |
Alias number line in Table1 |
GENBANK
Identification |
Date |
Number of 17711 UA/CG metastructures |
|
SARS-CoV2 Wuhan |
|
NC_045512.2 |
18-JUL-2020 |
8 |
|
INDIAA1 |
INDIAA1
MT415320 line1 |
MT415320.1 |
30-APR-2020 |
23 |
|
INDIAA2 |
INDIAA2
MT415321 line2 |
MT415321.1 |
30-APR-2020 |
8 |
|
INDIAA3 |
INDIAA3
MT415322 line3 |
MT415322.1 |
30-APR-2020 |
33 |
|
INDIAA4 |
INDIAA4
MT415323 line4 |
MT415323.1 |
30-APR-2020 |
45 |
|
INDIAA5 |
INDIAA5
MT457402 line6 |
MT457402.1 |
12-MAY-2020 |
45 |
|
INDIAA6 |
INDIAA5
MT457402 line7 |
MT457403.1 |
12-MAY-2020 |
34 |
|
INDIAA7 |
INDIA7
MT477885 line5 |
MT477885.1 |
18-MAY-2020 |
37 |
Genome’s lengths:
VSARSCOV2REF 29903
VINDIAA1 29900
VINDIAA2 29903
VINDIAA3 29888
VINDIAA4 29890
VINDIAA5 29890
VINDIAA6 29890
VINDIAA7 29899
6 of the 7 cases have deletions.
Only INDIAA2 has the same length as SARS-CoV2 Wuhan reference.
VSARSCOV2REF 29903
VINDIAA2 29903
Only 4 difference bases: it is precisely the only one that has not increased the number of metastructures.
Number of diferent bases: +/VSARSCOV2REF¬VINDIAA2 = 4
Locations: (VSARSCOV2REF¬VINDIAA2)/¼½VINDIAA2
241 3037 14408 23403
Nucleotides values in SARS-CoV2 ref: (VSARSCOV2REF¬VINDIAA2)/VSARSCOV2REF
CCCA
Nucleotides values in VINDIAA2: (VSARSCOV2REF¬VINDIAA2)/VINDIAA2
TTTG
i.e., 3 out of 4 CG mutations ==> UA
From the results below I deduce that the deletions of 5 cases out of 6 studied cases contributed to considerably increase the UA / CG metastructures of 17711 bases.
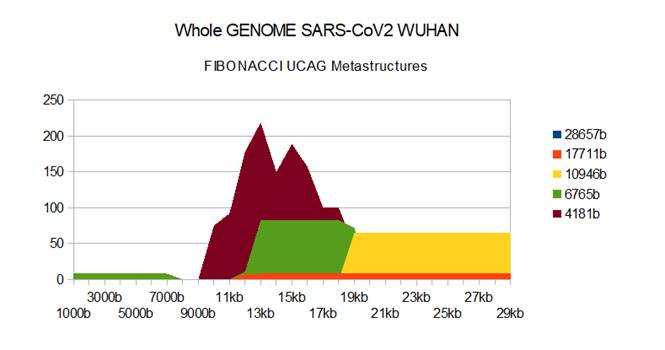
Figure 1: Recall SARS-CoV2 Wuhan genome metastructures.
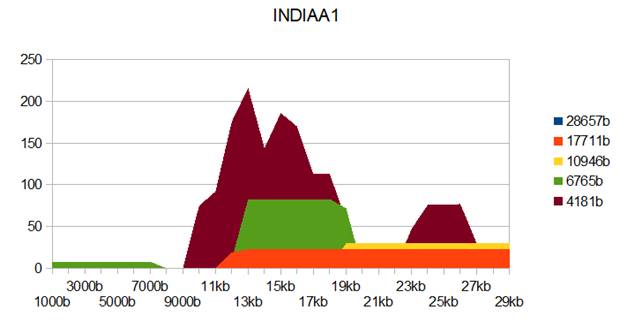
Figure 2: INDIAA1 genome metastructures.
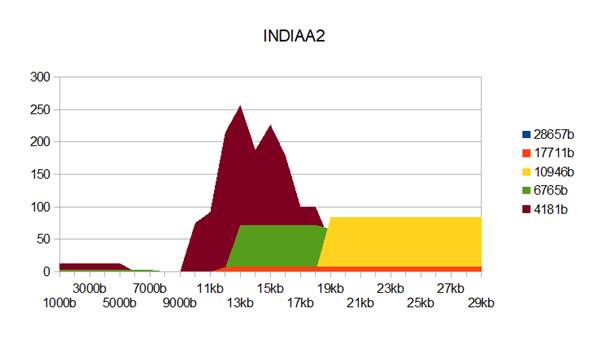
Figure 3: INDIAA2 genome metastructures.
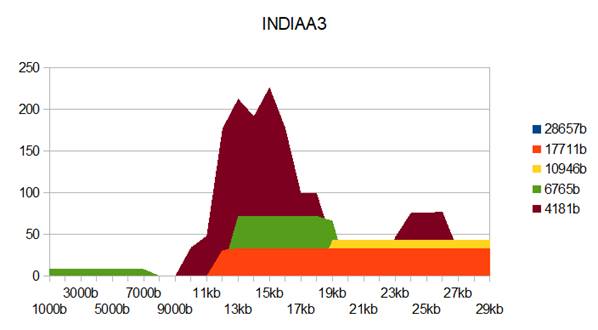
Figure 4: INDIAA3 genome metastructures.
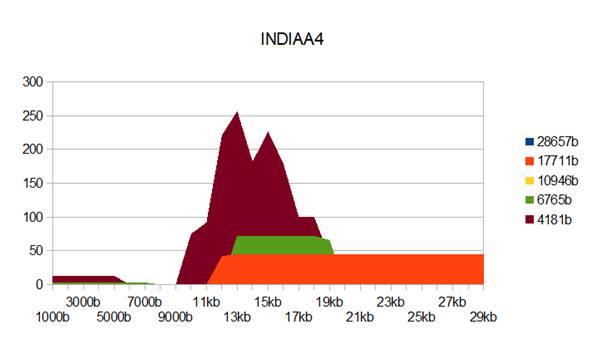
Figure 5: INDIAA4 genome metastructures.
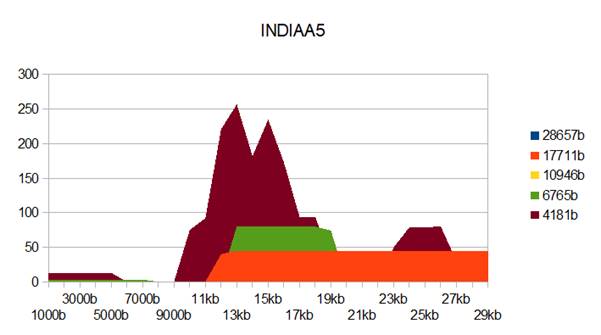
Figure 6: INDIAA5 genome metastructures.
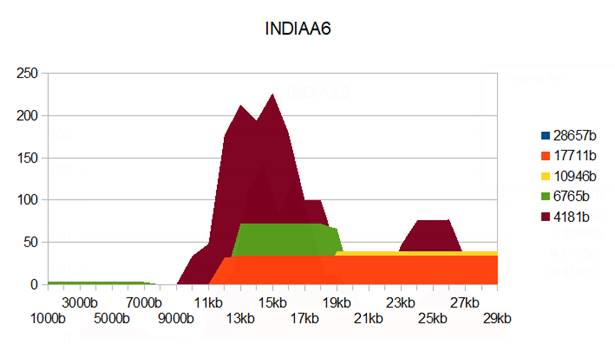
Figure 7: INDIAA6 genome metastructures.
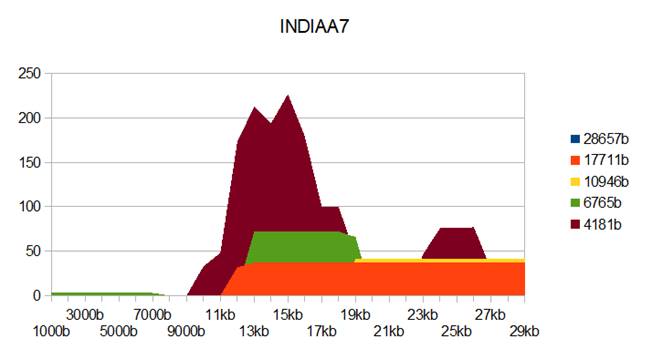
Figure 8: INDIAA7 genome metastructures.
We work now from these published data:
We test 2 possible variant scenarios:(Srivastava Surabhi et al, 2021), and more particularly on this Indian GEAR19 database: https://data.ccmb.res.in/gear19/variants
If separate mutations are INDIABn,
INDIACn, progressive descent by accumulating mutations by decreasing probabilities.
Example
INDIAC1 = INDIAB1
INDIAC2 = INDIAC1 + INDIAB2
INDIAC3 + INDIAC2 + INDIAB3 ...
…/...
INDIAC28 = INDIAC27 + INDIAB28
Then we study the most recent changes of
2021. For that we rely on this systematic approach:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7895735/
And more particularly on this Indian GEAR19 database:
https://data.ccmb.res.in/gear19/variants
link Table 5%:
On the table of 28 Indian mutations > 5% of cases, The progressive study of the 29 genomes by integrating mutations step by step according to their frequency should give very interesting Fibonacci on the scale of the whole genome.
INDIAC
INDIACn, progressive descent by accumulating mutations by decreasing probabilities.
Example INDIAC2 = INDIAC1 + INDIAB2
APL Language session mutations... ( ttps://en.wikipedia.org/wiki/APL_(programming_language ).
====== INDIAC =====
VINDIAC1„VSARSCOV2REF
VINDIAC1[23403] „'G'
VINDIAC2„VINDIAC1
VINDIAC2[3037] „'T'
VINDIAC3„VINDIAC2
VINDIAC3[241] „'T'
VINDIAC4„VINDIAC3
VINDIAC4[14408] „'T'
VINDIAC5„VINDIAC4
VINDIAC5[28881] „'A'
VINDIAC6„VINDIAC5
VINDIAC6[28883] „'C'
VINDIAC7„VINDIAC6
VINDIAC7[28882] „'A'
VINDIAC8„VINDIAC7
VINDIAC8[25563] „'T'
VINDIAC9„VINDIAC8
VINDIAC9[18877] „'T'
VINDIAC10„VINDIAC9
VINDIAC10[26735] „'T'
VINDIAC11„VINDIAC10
VINDIAC11[28854] „'T'
VINDIAC12„VINDIAC11
VINDIAC12[22444] „'T'
VINDIAC13„VINDIAC12
VINDIAC13[313] „'T'
VINDIAC14„VINDIAC13
VINDIAC14[5700] „'A'
VINDIAC15„VINDIAC14
VINDIAC15[11083] „'T'
VINDIAC16„VINDIAC15
VINDIAC16[13730] „'T'
VINDIAC17„VINDIAC16
VINDIAC17[28311] „'T'
VINDIAC18„VINDIAC17
VINDIAC18[23929] „'T'
VINDIAC19„VINDIAC18
VINDIAC19[6312] „'A'
VINDIAC20„VINDIAC19
VINDIAC20[8917] „'T'
VINDIAC21„VINDIAC20
VINDIAC21[1947] „'C'
VINDIAC22„VINDIAC21
VINDIAC22[9389] „'A'
VINDIAC23„VINDIAC22
VINDIAC23[6573] „'T'
VINDIAC24„VINDIAC23
VINDIAC24[4354] „'A'
VINDIAC25„VINDIAC24
VINDIAC25[25528] „'T'
VINDIAC26„VINDIAC25
VINDIAC26[15324] „'T'
VINDIAC27„VINDIAC26
VINDIAC27[3267] „'T'
VINDIAC28„VINDIAC27
VINDIAC28[3634] „'T'
Table 3: Summary on the 28 most frequent India country mutations applied to SARS-CoV2 Wuhan genome.
|
Position |
Genome location |
ref |
Alt |
gene |
Amino Acids mutations |
Percent |
Number of 17711 UA/CG metastructures |
|
SARS-CoV2 Wuhan |
|
|
|
|
|
|
8 |
|
INDIAC1 |
23403 |
A |
G |
"S :614" |
"D614G" |
85 |
8 |
|
INDIAC2 |
3037 |
C |
T |
"ORF1a :924" |
"F924F" |
84 |
8 |
|
INDIAC3 |
241 |
C |
T |
"5'UTR" |
"NA" |
84 |
8 |
|
INDIAC4 |
14408 |
C |
T |
"ORF1b :314" |
"P314L" |
84 |
8 |
|
INDIAC5 |
28881 |
G |
A |
"N :203" |
"R203K" |
42 |
31 |
|
INDIAC6 |
28883 |
G |
C |
"N :204" |
"G204R" |
41 |
31 |
|
INDIAC7 |
28882 |
G |
A |
"N :203" |
"R203K" |
40 |
46 |
|
INDIAC8 |
25563 |
G |
T |
"ORF3a :57" |
"Q57H" |
25 |
35 |
|
INDIAC9 |
18877 |
C |
T |
"ORF1b :1804" |
"L1804L" |
25 |
25 |
|
INDIAC10 |
26735 |
C |
T |
"M :71" |
"Y71Y" |
25 |
10 |
|
INDIAC11 |
28854 |
C |
T |
"N :194" |
"S194L" |
23 |
10 |
|
INDIAC12 |
22444 |
C |
T |
"S :294" |
"D294D" |
22 |
8 |
|
INDIAC13 |
313 |
C |
T |
"ORF1a :16" |
"L16L" |
21 |
8 |
|
INDIAC14 |
5700 |
C |
A |
"ORF1a :1812" |
"A1812D" |
20 |
8 |
|
INDIAC15 |
11083 |
G |
T |
"ORF1a :3606" |
"L3606F" |
14 |
8 |
|
INDIAC16 |
13730 |
C |
T |
"ORF1b :88" |
"A88V" |
11 |
29 |
|
INDIAC17 |
28311 |
C |
T |
"N :13" |
"P13L" |
10 |
29 |
|
INDIAC18 |
23929 |
C |
T |
"S :789" |
"Y789Y" |
10 |
41 |
|
INDIAC19 |
6312 |
C |
A |
"ORF1a :2016" |
"T2016K" |
10 |
41 |
|
INDIAC20 |
8917 |
C |
T |
"ORF1a :2884" |
"F2884F" |
10 |
41 |
|
INDIAC21 |
1947 |
T |
C |
"ORF1a :561" |
"V561A" |
7 |
41 |
|
INDIAC22 |
9389 |
G |
A |
"ORF1a :3042" |
"D3042N" |
6 |
41 |
|
INDIAC23 |
6573 |
C |
T |
"ORF1a :2103" |
"S2103F" |
6 |
41 |
|
INDIAC24 |
4354 |
G |
A |
"ORF1a :1363" |
"E1363E" |
6 |
41 |
|
INDIAC25 |
25528 |
C |
T |
"ORF3a :46" |
"L46F" |
6 |
48 |
|
INDIAC26 |
15324 |
C |
T |
"ORF1b :619" |
"N619N" |
6 |
36 |
|
INDIAC27 |
3267 |
C |
T |
"ORF1a :1001" |
"T1001I" |
6 |
36 |
|
INDIAC28 |
3634 |
C |
T |
"ORF1a :1123" |
"N1123N" |
6 |
36 |
|
Average |
|
|
|
|
|
26.25% |
26.89 |
Table 4: Recall summary main results from Table3.
|
Genome |
Percent % |
Number of 17711 UA/CG metastructures |
|
SARS-CoV2 Wuhan |
None |
8 |
|
INDIAC1 |
85 |
8 |
|
INDIAC2 |
84 |
8 |
|
INDIAC3 |
84 |
8 |
|
INDIAC4 |
84 |
8 |
|
INDIAC5 |
42 |
31 |
|
INDIAC6 |
41 |
31 |
|
INDIAC7 |
40 |
46 |
|
INDIAC8 |
25 |
35 |
|
INDIAC9 |
25 |
25 |
|
INDIAC10 |
25 |
10 |
|
INDIAC11 |
23 |
10 |
|
INDIAC12 |
22 |
8 |
|
INDIAC13 |
21 |
8 |
|
INDIAC14 |
20 |
8 |
|
INDIAC15 |
14 |
8 |
|
INDIAC16 |
11 |
29 |
|
INDIAC17 |
10 |
29 |
|
INDIAC18 |
10 |
41 |
|
INDIAC19 |
10 |
41 |
|
INDIAC20 |
10 |
41 |
|
INDIAC21 |
7 |
41 |
|
INDIAC22 |
6 |
41 |
|
INDIAC23 |
6 |
41 |
|
INDIAC24 |
6 |
41 |
|
INDIAC25 |
6 |
48 |
|
INDIAC26 |
6 |
36 |
|
INDIAC27 |
6 |
36 |
|
INDIAC28 |
6 |
36 |
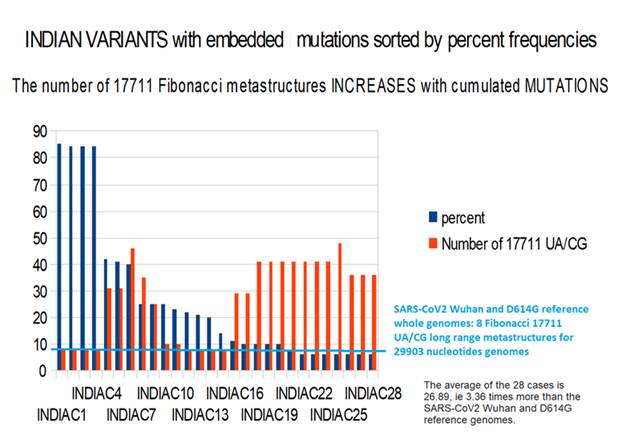
Figure 9: Increase of 17711 UA/CG metastructures with whole INDIAN variant genomes with cumulated mutations vs percent frequencies (vs SARS-CoV2 Wuhan).
From this analysis, we can draw 3 conclusions:
1) this is a simulation of genomes made from SARS-CoV2 and the most frequently encountered mutations in India. So, if it is certain that the first genomes exist in some patients, some others, towards the end of the list of 28 genomes, may not exist but could potentially emerge.
2) it is noted that none of the 28 cases found UA / CG metastructures of 177122 bases in quantity LESS than 8, a value which characterizes SARS-CoV2 Wuhan.
So, if there was no correlation between these Fibonacci metastructures and the evolution of variants, we should find cases less than 8.
3) out of the 28 cases of genomes studied, 20 of them saw an increase in the number of metastructures of 17,712 bases, or more than 2/3 of the genomes studied. The average of the 28 cases is 26.89, ie 3.36 times more than the SARS-CoV2 Wuhan and D614G reference genomes.
The strain of the variant B.1.617 has grown exponentially in India since the beginning of 2021. We are going to redo the 28 previous analyzes no longer from the SARS-CoV2 Wuhan genome but by inserting the SINDIAFULL spike already analyzed in (Perez, 2021).
This therefore amounts to applying the successive mutations to a type B 1.617 genome, at least at the level of its Spike sequence.
Indeed,
B.1.617 lineage
This strain, also known as the “double mutant virus”, has spread rapidly through India.
The strain has been dubbed the “double mutant virus” due to two of the concerning mutations it carries.
These two key mutations are:
E484Q
L452R
Further studies on the strain are needed to determine its transmissibility, although it is suspected to do so due to its spike protein mutations which are thought to increase immune evasion and receptor binding. Whether vaccine efficacy is affected also needs further research.
SINDIAFULL is the Spike B.1.617 from (Perez-2021).
Recall Spike location
21563..25384
/gene="S"
APL Language session mutations... ( ttps://en.wikipedia.org/wiki/APL_(programming_language) ).
½V„VSARSCOV2REF [21562+¼ (25384-21562)]
3822
V[¼9]
ATGTTTGTT
½SINDIAFULL
3822
SINDIAFULL[¼9]
ATGTTTGTT
VB1617„VSARSCOV2REF
½VB1617[21562+¼ (25384-21562)] „SINDIAFULL
3822
+/VB1617¬VSARSCOV2REF
8
(VB1617¬VSARSCOV2REF)/VSARSCOV2REF
GAGGTGAA
(VB1617¬VSARSCOV2REF)/VB1617
TCTTGATG
R„GFIBOZOOMS VB1617
VINDIAC1„VB1617
VINDIAC1[23403] „'G'
VINDIAC2„VINDIAC1
VINDIAC2[3037] „'T'
VINDIAC3„VINDIAC2
VINDIAC3[241] „'T'
VINDIAC4„VINDIAC3
VINDIAC4[14408] „'T'
VINDIAC5„VINDIAC4
VINDIAC5[28881] „'A'
VINDIAC6„VINDIAC5
VINDIAC6[28883] „'C'
VINDIAC7„VINDIAC6
VINDIAC7[28882] „'A'
VINDIAC8„VINDIAC7
VINDIAC8[25563] „'T'
VINDIAC9„VINDIAC8
VINDIAC9[18877] „'T'
VINDIAC10„VINDIAC9
VINDIAC10[26735] „'T'
VINDIAC11„VINDIAC10
VINDIAC11[28854] „'T'
VINDIAC12„VINDIAC11
VINDIAC12[22444] „'T'
VINDIAC13„VINDIAC12
VINDIAC13[313] „'T'
VINDIAC14„VINDIAC13
VINDIAC14[5700] „'A'
VINDIAC15„VINDIAC14
VINDIAC15[11083] „'T'
VINDIAC16„VINDIAC15
VINDIAC16[13730] „'T'
VINDIAC17„VINDIAC16
VINDIAC17[28311] „'T'
VINDIAC18„VINDIAC17
VINDIAC18[23929] „'T'
VINDIAC19„VINDIAC18
VINDIAC19[6312] „'A'
VINDIAC20„VINDIAC19
VINDIAC20[8917] „'T'
Table 5: Summary on the 28 most frequent India country mutations applied to B.1.617 genome.
|
Position |
Genome location |
ref |
Alt |
gene |
Amino Acids mutations |
Percent |
Number of 17711 UA/CG metastructures |
|
SARS-CoV2 Wuhan |
|
|
|
|
|
|
8 |
|
B1617 |
|
|
|
|
|
|
31 |
|
INDIAC1 |
23403 |
A |
G |
"S :614" |
"D614G" |
85 |
31 |
|
INDIAC2 |
3037 |
C |
T |
"ORF1a :924" |
"F924F" |
84 |
31 |
|
INDIAC3 |
241 |
C |
T |
"5'UTR" |
"NA" |
84 |
31 |
|
INDIAC4 |
14408 |
C |
T |
"ORF1b :314" |
"P314L" |
84 |
46 |
|
INDIAC5 |
28881 |
G |
A |
"N :203" |
"R203K" |
42 |
35 |
|
INDIAC6 |
28883 |
G |
C |
"N :204" |
"G204R" |
41 |
35 |
|
INDIAC7 |
28882 |
G |
A |
"N :203" |
"R203K" |
40 |
25 |
|
INDIAC8 |
25563 |
G |
T |
"ORF3a :57" |
"Q57H" |
25 |
10 |
|
INDIAC9 |
18877 |
C |
T |
"ORF1b :1804" |
"L1804L" |
25 |
10 |
|
INDIAC10 |
26735 |
C |
T |
"M :71" |
"Y71Y" |
25 |
8 |
|
INDIAC11 |
28854 |
C |
T |
"N :194" |
"S194L" |
23 |
29 |
|
INDIAC12 |
22444 |
C |
T |
"S :294" |
"D294D" |
22 |
29 |
|
INDIAC13 |
313 |
C |
T |
"ORF1a :16" |
"L16L" |
21 |
29 |
|
INDIAC14 |
5700 |
C |
A |
"ORF1a :1812" |
"A1812D" |
20 |
29 |
|
INDIAC15 |
11083 |
G |
T |
"ORF1a :3606" |
"L3606F" |
14 |
29 |
|
INDIAC16 |
13730 |
C |
T |
"ORF1b :88" |
"A88V" |
11 |
41 |
|
INDIAC17 |
28311 |
C |
T |
"N :13" |
"P13L" |
10 |
48 |
|
INDIAC18 |
23929 |
C |
T |
"S :789" |
"Y789Y" |
10 |
36 |
|
INDIAC19 |
6312 |
C |
A |
"ORF1a :2016" |
"T2016K" |
10 |
36 |
|
INDIAC20 |
8917 |
C |
T |
"ORF1a :2884" |
"F2884F" |
10 |
36 |
|
INDIAC21 |
1947 |
T |
C |
"ORF1a :561" |
"V561A" |
7 |
36 |
|
INDIAC22 |
9389 |
G |
A |
"ORF1a :3042" |
"D3042N" |
6 |
36 |
|
INDIAC23 |
6573 |
C |
T |
"ORF1a :2103" |
"S2103F" |
6 |
36 |
|
INDIAC24 |
4354 |
G |
A |
"ORF1a :1363" |
"E1363E" |
6 |
36 |
|
INDIAC25 |
25528 |
C |
T |
"ORF3a :46" |
"L46F" |
6 |
34 |
|
INDIAC26 |
15324 |
C |
T |
"ORF1b :619" |
"N619N" |
6 |
62 |
|
INDIAC27 |
3267 |
C |
T |
"ORF1a :1001" |
"T1001I" |
6 |
62 |
|
INDIAC28 |
3634 |
C |
T |
"ORF1a :1123" |
"N1123N" |
6 |
62 |
|
Average |
|
|
|
|
|
26.25% |
34.57 |
Table 6: Recall summary main results from Table5.
|
Genome |
Percent % |
Number of 17711 UA/CG metastructures |
|
SARS-CoV2 Wuhan |
None |
8 |
|
B1.617 |
|
31 |
|
INDIAC1 |
85 |
31 |
|
INDIAC2 |
84 |
31 |
|
INDIAC3 |
84 |
31 |
|
INDIAC4 |
84 |
46 |
|
INDIAC5 |
42 |
35 |
|
INDIAC6 |
41 |
35 |
|
INDIAC7 |
40 |
25 |
|
INDIAC8 |
25 |
10 |
|
INDIAC9 |
25 |
10 |
|
INDIAC10 |
25 |
8 |
|
INDIAC11 |
23 |
29 |
|
INDIAC12 |
22 |
29 |
|
INDIAC13 |
21 |
29 |
|
INDIAC14 |
20 |
29 |
|
INDIAC15 |
14 |
29 |
|
INDIAC16 |
11 |
41 |
|
INDIAC17 |
10 |
48 |
|
INDIAC18 |
10 |
36 |
|
INDIAC19 |
10 |
36 |
|
INDIAC20 |
10 |
36 |
|
INDIAC21 |
7 |
36 |
|
INDIAC22 |
6 |
36 |
|
INDIAC23 |
6 |
36 |
|
INDIAC24 |
6 |
36 |
|
INDIAC25 |
6 |
34 |
|
INDIAC26 |
6 |
62 |
|
INDIAC27 |
6 |
62 |
|
INDIAC28 |
6 |
62 |
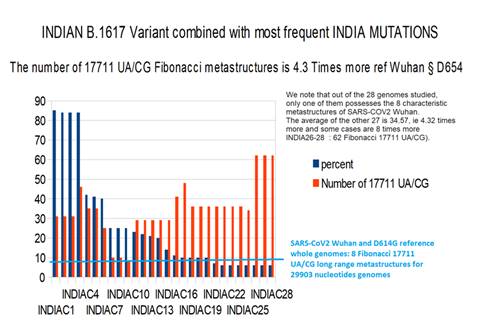
Figure 10: Increase of 17711 UA/CG metastructures with whole INDIAN variant genomes with cumulated mutations vs percent frequencies (vs. B.1.617 variant).
The most remarkable result is the fact that the very simple combination of the 4 most frequent mutations (85% of cases) and the variant B.1.617 is sufficient to multiply by 4 to 6 (31 to 46 against 8 for SARS-CoV2 Wuhan (the number of Fibonacci metastructures of 17,712 AU / CG bases. We also note that out of the 28 genomes studied, only one of them possesses the 8 characteristic metastructures of SARS-COV2 Wuhan. The average of the other 27 is 34.57, ie 4.32 times more and some cases are 8 times more INDIA26-28: 62).
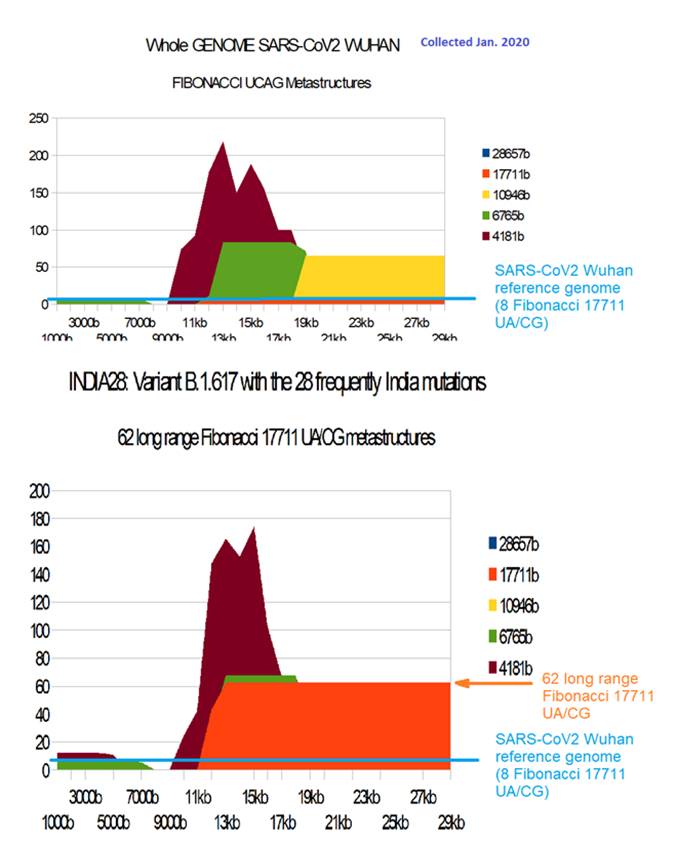
Figure 11: Comparing long range 17711 UA/CG Fibonacci metastructures between SARS-CoV2 Wuhan and India variant B.1.617 with the 28 most frequent India country mutations.
In (Pragya Yadav et al, 2021), the authors provide a list of the 33 main mutations characterizing the genomes of the Indian variant B 1.617.
On the other hand, we have just studied the impact of the 28 most frequent mutations in India, those which represent more than 5% of contaminations).
It is clear that these 2 sets of mutations partially overlap.
However, it would be interesting to simulate the effect of some of the 28 mutations when they are absent in B.1.617. Indeed, their high frequency makes it possible to suggest their possible future addition to B.1.617.
This is what we will simulate in this last paragraph.
Table 7: The 33 main muatations from India variant B.1.617 from (Pragya Yadav et al., 2021). From Figure 1: nCharacteristics and neutralization of VUI B.1.617 variant: A) nThe common nucleotide changes observed in majority of the isolates and clinical sequences. We identify 22 other frequent mutations in India (frequency greater than 5% of contaminations) but absent in the Indian variant B.1.617.
Note: We rename these successive mutants SINDIA6 for B.1.617 consensus + INDIA6 etc ...
In this Table 7, there are 2 parts:
-In the top part, the 32 mutations characterizing the India variant B.1.617 REF.
-In the bottom part, we run the 22 remaining mutations from the 28 most frequent mutations in full India country. Then, we test two cases: cumulating the 22 successive mutations, then running each mutation separately. In all cases, the amount of 17711 UA/CG Fibonacci metastructures is around 5 (« Ten ») times the number of 17711 UA/CG in SARS-CoV2 Wuhan reference genome.
Analysing the 32 mutations concensus India variant B.617:
We run here the 32 mutations applied to SARS-CoV2 reference Wuhan genome:
APL Language session mutations... (ttps://en.wikipedia.org/wiki/APL_(programming_language)).
B1617REF = VSARSCOV2REF
B1617REF [210]
B1617REF [210] „'T'
B1617REF [241]
C
B1617REF [241] „'T'
B1617REF [3037]
C
B1617REF [3037] „'T'
B1617REF [3457]
C
B1617REF [3457] „'T'
B1617REF [4965]
C
B1617REF [4965] „'T'
B1617REF [8491]
G
B1617REF [8491] „'A'
B1617REF [11201]
A
B1617REF [11201] „'G'
B1617REF [14408]
C
B1617REF [14408] „'T'
B1617REF [14772]
G
B1617REF [14772] „'A'
B1617REF [16134]
C
B1617REF [16134] „'T'
B1617REF [16852]
G
B1617REF [16852] „'T'
B1617REF [17523]
G
B1617REF [17523] „'T'
B1617REF [20396]
A
B1617REF [20396] „'G'
B1617REF [20401]
T
B1617REF [20401] „'G'
B1617REF [21846]
C
B1617REF [21846] „'T'
B1617REF [21895]
T
B1617REF [21895] „'C'
B1617REF [21987]
G
B1617REF [21987] „'A'
B1617REF [22022]
G
B1617REF [22022] „'A'
B1617REF [22917]
T
B1617REF [22917] „'G'
B1617REF [23012]
G
B1617REF [23012] „'C'
B1617REF [23403]
A
B1617REF [23403] „'G'
B1617REF [23604]
C
B1617REF [23604] „'G'
B1617REF [24775]
A
B1617REF [24775] „'T'
B1617REF [25469]
C
B1617REF [25469] „'T'
B1617REF [26767]
T
B1617REF [26767] „'G'
B1617REF [27382]
G
B1617REF [27382] „'C'
B1617REF [27385]
T
B1617REF [27385] „'C'
B1617REF [27520]
A
B1617REF [27520] „'T'
B1617REF [27638]
T
B1617REF [27638] „'C'
B1617REF [28881]
G
B1617REF [28881] „'T'
B1617REF [29402]
G
B1617REF [29402] „'T'
B1617REF [29742]
G
B1617REF [29742] „'T'
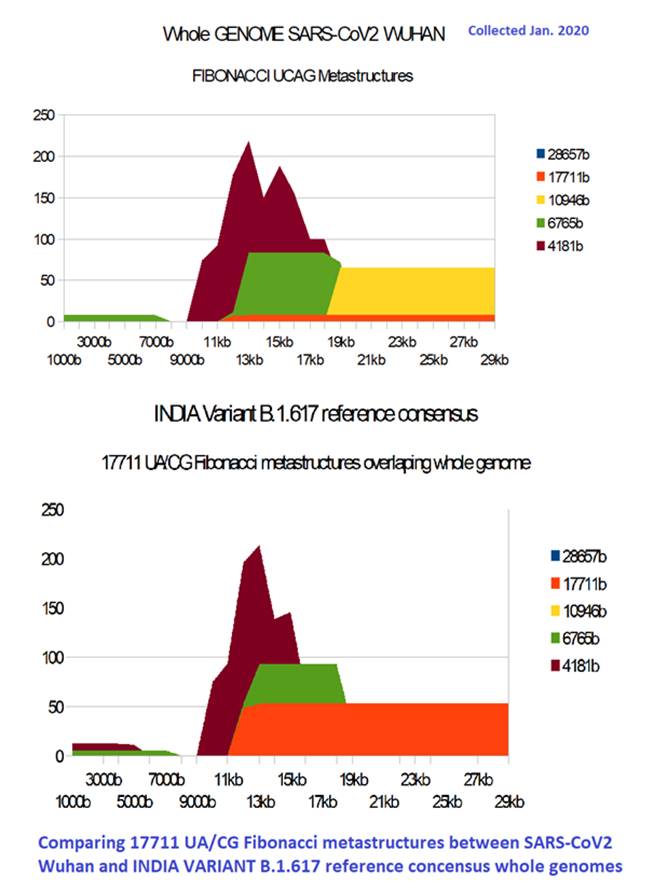
Figure 12: Comparing long range 17711 UA/CG Fibonacci metastructures between SARS-CoV2 Wuhan and India variant B.1.617 Reference Concensus (Pragya Yadav et al, 2021) including 32 mutations.
Now we will apply to this strain B.1.617 consensus the progressive accumulation of the 22 other frequent mutations in India (frequency greater than 5% of contaminations) but absent in the Indian variant B.1.617.
For this purpose, as we did in the previous §, we will apply to B.1.617 consensus each of the 22 mutations, accumulating them one by one and respecting the order of their frequency of contamination in India (here in the order INDIAC6, then INDIAC6 + INDIAC7, then INDIAC6 + INDIAC7 + INDIAC8 ... as these muattions appear in Table 7.
Note: We rename these successive mutants SINDIA6 for B.1.617 consensus + INDIA6 etc...
APL Language session mutations... (ttps://en.wikipedia.org/wiki/APL_(programming_language)).
SINDIAC6„B1617REF
SINDIAC6[28883]
G
SINDIAC6[28883] „'C'
SINDIAC7„SINDIAC6
SINDIAC7[28882]
G
SINDIAC7[28882] „'A'
SINDIAC8„SINDIAC7
SINDIAC8[25563]
G
SINDIAC8[25563] „'T'
SINDIAC10„SINDIAC8
SINDIAC10[26735]
C
SINDIAC10[26735] „'T'
SINDIAC11„SINDIAC10
SINDIAC11[28854]
SINDIAC11[28854] „'T'
SINDIAC12„SINDIAC11
SINDIAC12[22444]
C
SINDIAC12[22444] „'T'
SINDIAC13„SINDIAC12
SINDIAC13[313]
C
SINDIAC13[313] „'T'
SINDIAC14„SINDIAC13
SINDIAC14[5700]
C
SINDIAC14[5700] „'A'
SINDIAC15„SINDIAC14
SINDIAC15[11083]
G
SINDIAC15[11083] „'T'
SINDIAC16„SINDIAC15
SINDIAC16[13730]
C
SINDIAC16[13730] „'T'
SINDIAC17„SINDIAC16
SINDIAC17[23929]
C
SINDIAC17[28311] „'T'
SINDIAC18„SINDIAC17
SINDIAC18[23929]
C
SINDIAC18[23929] „'T'
SINDIAC19„SINDIAC18
SINDIAC19[6312]
C
SINDIAC19[6312] „'A'
SINDIAC20„SINDIAC19
SINDIAC20[8917]
C
SINDIAC20[8917] „'T'
SINDIAC21„SINDIAC20
SINDIAC21[1947]
T
SINDIAC21[1947] „'C'
SINDIAC22„SINDIAC21
SINDIAC22[9389]
G
SINDIAC22[9389] „'A'
SINDIAC23„SINDIAC22
SINDIAC23[6573]
C
SINDIAC23[6573] „'T'
SINDIAC24„SINDIAC23
SINDIAC24[4354]
G
SINDIAC24[4354] „'A'
SINDIAC25„SINDIAC24
SINDIAC25[25528]
C
SINDIAC25[25528] „'T'
SINDIAC26„SINDIAC25
SINDIAC26[15324]
C
SINDIAC26[15324] „'T'
SINDIAC27„SINDIAC26
SINDIAC27[3267]
C
SINDIAC27[3267] „'T'
SINDIAC28„SINDIAC27
SINDIAC28[3634]
C
SINDIAC28[3634] „'T'
Here, unlike the 2 previous simulations where most of the mutations INCREASED the number of long AU / CG metastructures, here almost all of the mutations DECREASE the number of these long metastructures. It is true that the level of these metastructures of 17711 AU / CG bases is very IMPORTANT in the reference genome B.1.617 Ref.
The level of the B.1.617 consensus
reference variant genome is however more than 6.6 times higher than that of the
Wuhan SARS-CoV2 reference genome.
The average level of these 22 nested mutations applied to the variant genome consensus reference B.1.617 is however more than 4.7 times higher than that of the reference genome SARS-CoV2 Wuhan.
See results in Figure 13 below.
What about running the same 22 mutations
but with INDIVIDUAL MUTATIONS instead of cumulated mutations?
APL Language session mutations... (ttps://en.wikipedia.org/wiki/APL_(programming_language)).
MEMO22INDIVIDUALS
SINDIAC6„B1617REF
SINDIAC7„B1617REF
SINDIAC8„B1617REF
SINDIAC10„B1617REF
SINDIAC11„B1617REF
SINDIAC12„B1617REF
SINDIAC13„B1617REF
SINDIAC14„B1617REF
SINDIAC15„B1617REF
SINDIAC16„B1617REF
SINDIAC17„B1617REF
SINDIAC18„B1617REF
SINDIAC19„B1617REF
SINDIAC20„B1617REF
SINDIAC21„B1617REF
SINDIAC22„B1617REF
SINDIAC23„B1617REF
SINDIAC24„B1617REF
SINDIAC25„B1617REF
SINDIAC26„B1617REF
SINDIAC27„B1617REF
SINDIAC28„B1617REF
SINDIAC6[28883] „'C'
SINDIAC7[28882] „'A'
SINDIAC8[25563] „'T'
SINDIAC10[26735] „'T'
SINDIAC11[28854] „'T'
SINDIAC12[22444] „'T'
SINDIAC13[313] „'T'
SINDIAC14[5700] „'A'
SINDIAC15[11083] „'T'
SINDIAC16[13730] „'T'
SINDIAC17[28311] „'T'
SINDIAC18[23929] „'T'
SINDIAC19[6312] „'A'
SINDIAC20[8917] „'T'
SINDIAC21[1947] „'C'
SINDIAC22[9389] „'A'
SINDIAC23[6573] „'T'
SINDIAC24[4354] „'A'
SINDIAC25[25528] „'T'
SINDIAC26[15324] „'T'
SINDIAC27[3267] „'T'
SINDIAC28[3634] „'T'
Table 8: Evolution of 17711 UA/CG metastructures with whole INDIAN variant genomes with CUMULATED then SEPARATED mutations vs percent frequencies (vs. B.1.617 REF variant).
|
Genome |
Percent % |
CUMULATING 22 Mutations : 17711 UA/CG
Fibonacci metastructures |
SEPARATE 22 Mutations : 17711 UA/CG
Fibonacci metastructures |
|
SARS-CoV2 Wuhan |
None |
8 |
8 |
|
B1.617 reference |
|
53 |
|
|
28883 SINDIAC6 |
41 |
53 |
53 |
|
28882 SINDIAC7 |
40 |
32 |
32 |
|
25563 SINDIAC8 |
25 |
21 |
32 |
|
26735 SINDIAC10 |
25 |
14 |
32 |
|
28854 SINDIAC11 |
23 |
8 |
32 |
|
22444 SINDIAC12 |
22 |
31 |
32 |
|
313 SINSDIAC13 |
21 |
31 |
53 |
|
5700 SINDIAC14 |
20 |
31 |
53 |
|
11083 SINDIAC15 |
14 |
31 |
53 |
|
13730 SINDIAC16 |
11 |
28 |
32 |
|
28311 SINDIAC17 |
10 |
40 |
32 |
|
23929 SINDIAC18 |
10 |
48 |
32 |
|
6312 SINDIAC19 |
10 |
48 |
53 |
|
8917 SINDIAC20 |
10 |
48 |
53 |
|
1947 SINDIAC21 |
7 |
48 |
53 |
|
9389 SINDIAC22 |
6 |
48 |
53 |
|
6573 SINDIAC23 |
6 |
48 |
53 |
|
4354 SINDIAC24 |
6 |
48 |
53 |
|
25528 SINDIAC25 |
6 |
38 |
32 |
|
15324 SINDIAC26 |
6 |
45 |
32 |
|
3267 SINDIAC27 |
6 |
45 |
53 |
|
3634 SINDIAC28 |
6 |
45 |
53 |
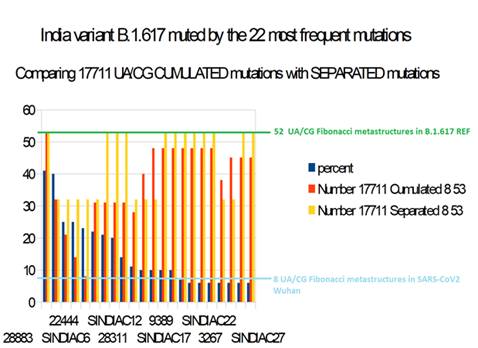
Figure 13: Evolution of 17711 UA/CG metastructures with whole INDIAN variant genomes with CUMULATED (red) and SEPARETED (yellow) mutations vs percent frequencies (vs. B.1.617 REF variant).
Globally, all simulated whole genomes have a number of 17711 UA/CG metastructures greater than the initial SARS-CoV2 Wuhan genome.
In the spring of 2021, an endogenous strain of the Indian variant B.1.617 developed exponentially in India, then in England, then it was reported in about fifty countries, it is the dominant strain B.1.617.2.
We must recall propertie of the 3 B.1.617 substrains:
B.1.617.1 detected Dec 2020
B.1.617.2 detected Dec 2020
B.1.617.3 detected Oct 2020
B.1.617 contains 3 clades with different mutation profiles which are:
• B.1.617.1 – includes a large number of sequences and has a spike profile including L452R and E484Q.
• B.1.617.2 – has a different profile without E484Q and appears to have recent
expansion.
• B.1.617.3 – has L452R and E484Q but is distinct from B.1.617.1 and currently remains small.
What differentiates this variant, both in terms of the entire genome and its spike, compared to the Wuhan or D614G reference strains?
Here is the list of the various mutations of this variant (source www.covariants.org).
20A/S:478K is also known as B.1.617.2

20A/S:154K

Defining mutations


Comparing B.1.617.2 Spike with Wuhan
original Spike:
In Figure 14, we do not notice any big differences between the respective profiles of the UA / CG metastructures of these 2 Spikes. In particular, the 2584 AU / CG metastructures remain weak (blue), the 1597 AU / CG metastructures (red) retain their remarkable "podium" structure. On the other hand, in B.1.617.2 Spike, appear two sharp points characterizing the 987 AU / CG (yellow).
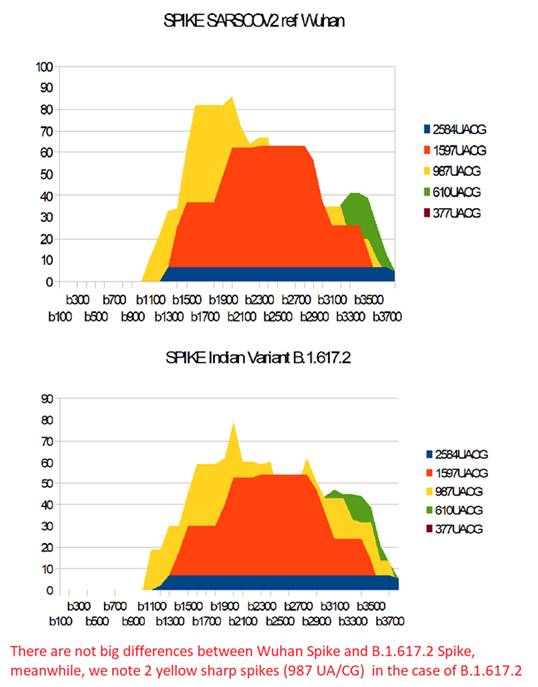
Figure 14: Comparing SARS-CoV2 Wuhan and B.1.617.2 India variant mRNA Spikes.
Comparing B.1.617.2 whole Genome with
Wuhan original whole Genome:
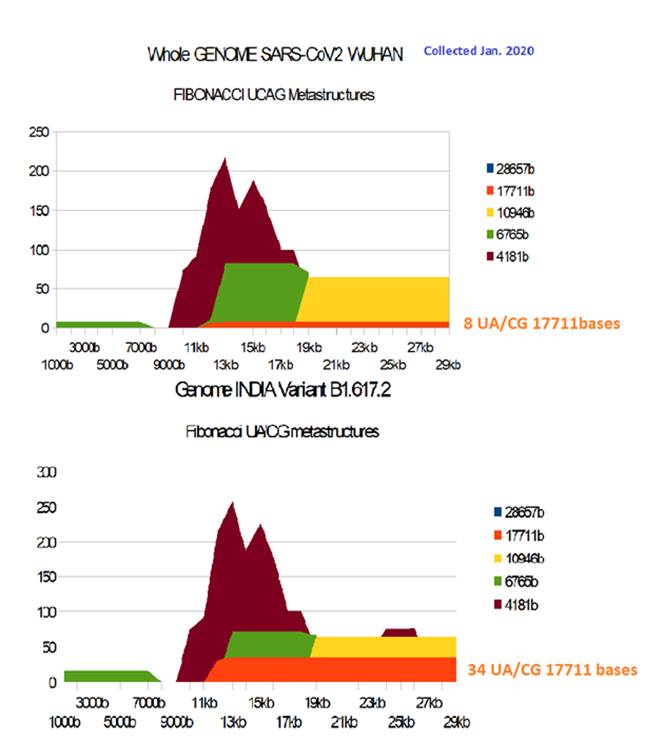
Figure 15: Comparing SARS-CoV2 Wuhan and B.1.617.2 India variant whole genomes.
Summary of this study.
Unlike the Spikes for which the UA / CG metastructures hardly differentiated the Wuhan and B.1.617.2 strains, here, at the scale of whole genomes (Figure 15), we find that the very long metastructures of 17711 UA / CG are multiplied by more than 4 times (8 ==> 34) between the Wuhan and B.1.617.2 genomes.
As demonstrated by Luc Montagnier from 1963 (Montagnier L. et al., 1963) And as he specified in May 2021 in this video about the stability of the SARS-CoV2 mRNA (Montagnier L., 2021), let us specify that the mRNA of a virus quickly transforms into a double chain of DNA of considerable strength.
We suggest that these very dense and long Fibonacci metastructures precisely reinforce the strength and the lifespan of the fragile mRNA of the virus, but also of the resulting DNA.
Recall, as we note in (Perez jc, 2021) that these Fibonacci metastructures are observed in all strains and variants of SARS-CoV2 while they are completely absent in the mRNAs of the spikes (modified at the level of synonymous codons) carried by the mRNA vaccines from Pfizer and Moderna.
Indeed, in order to maximize their mRNA stability, these 2 vaccines were overloaded with G bases. mRNA, thus also of their DNA, and probably also a low production of antibodies.
In the first part of this study, we limit ourselves to the analysis of whole genomes, all coming from the mutations and variants of SARS-CoV2 sequenced in India in 2020 and 2021. We then demonstrate - both on actual genomes of patients and on variants combining the most frequent mutations to the SARS-CoV2 Wuhan genomes and then to the B.1.617 variant - that the numerical Fibonacci AU / CG metastructures increase considerably in all cases analyzed in ratios of up to 8 times. We can affirm that this property contributes to a greater stability and lifespan of messenger RNAs, therefore, possibly also to a greater INFECTUOSITY of these variant genomes.
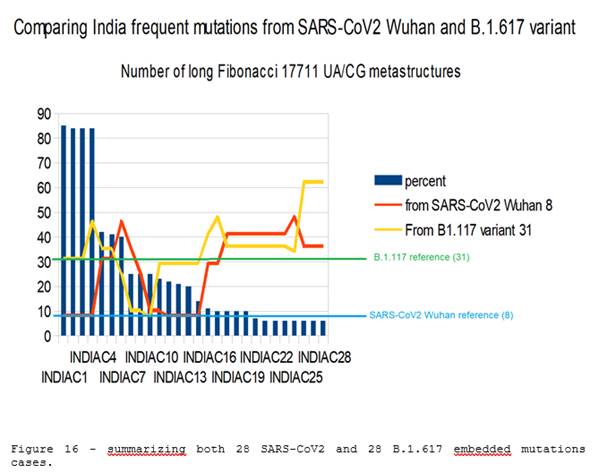
Figure 16: summarizing both 28 SARS-CoV2 and 28 B.1.617 embedded mutations cases.
In this study, we looked for the presence and number of UA / CG Fibonacci metastructures. We are interested in the longest of 17,711 bases, for genomes of 29,000 bases. These genomes concerned, for some, real patients, and, for others, the 28 mutations and variants most frequent in India, those which represent more than 5% of the cases of infections of the country.
Out of a total of 108 genomes analyzed:
· None ("NONE") of them contained a number of metastructures LOWER than those of the reference SARS-CoV2 Wuhan genome.
· Eleven (11) among them contained the same number of metastructures as the reference genome.
· 97 of them contained a GREATER number of metastructures than the reference genome, ie 89.81% of cases. The average increase in the number of metastructures for the 97 cases studied is 4.35 times the number of SARS-CoV2 UA/CG 17711 Fibonacci metastructures.( 4.35 = 34.83 / 8 ).
Note the impact of the new B.1.617 variant which, combined with the 4 most frequent mutations (85% of contaminations in the country), multiplies by 4 ("four") the number of metastructures of 17,711 bases compared to the reference genome SARS-CoV2 (8 ==> 31). It is therefore clear that the evolutionary pressure of mutations and variants operates on the mRNAs of viruses a sort of adaptation and even OPTIMIZATION of the AU / CG ratios of the entire genome. Only the virus "knows" this STRATEGY, and we think we have unveiled a corner of the veil here …
When we run the most frequent mutations
in India whole country, on the reference consensus B.1.617 India Variant, the
level of the B.1.617 consensus reference variant genome is more than 6.6 times
(53+8) higher than that of the Wuhan SARS-CoV2 reference genome.
Now, an open question:
Is there a SARSCOV2 Variants Evolution
Global Strategy?
To demonstrate a hypothetical global variant strategy, we have gathered 19 variants representative of the great diversity of variants:
-bat RaTG13, reputed to be very close to SARS-CoV2.
-The 2 original strains Wuhan and D614G.
-A strain related to mink (Hammer et al., 2021).
A strain Marseille4, including 13 mutations, which according to professor Didier
Raoult, coming from Africa, close to mink strains, was in the majority in this region of France before being erased by the English variant (Fournier et al., 2021).
-the 3 English variants.
-the 4 South African variants.
-the 3 Brazilian variants.
-the Californian variant Cal20. C.
-the 2 Indian variants B.1.617 and B.1.617.2.
When we compare the Fibonacci of these 19 spikes, it appears (Table 9 and Figure 17) that the majority of the variants see their longest metastructure 2584 AU / CG almost always greater than much greater than that of the spikes of the 2 reference genomes.
However, a low value is noted for Marseille4, reputed to be excessively pathogenic.
We also note a low value for Mink, whose codon reading frame is shifted shortly after the PRRA insertion point.
On the contrary, bat RaTG13 is characterized by a very high value (40 against 6 for Wuhan Spike).
The analysis of "podium like" 1597 AU / CG is very interesting because it highlights strong imbalances (LEFT California, or RIGHT Marseille4) between the left and right parts of the podium, this reflects an imbalance of Fibonacci 1597bases between the regions S1 and S2 of the Spike, we suggest that this imbalance may be associated with greater PATHOGENICITY.
On the contrary, several variants are located in a region of equilibrium between the 2 left and right parts of the podium, this is the case of the 3 English variants but also despite a slight imbalance of the Indian variant B.1.617 2. We suggest that these equilibria induce greater INFECTUOSITY.
Table 9: Comparing 19 SARS-CoV2 variants 2584 UA/CG Spike Fibonacci metastructures.
|
SPIKES |
Long range Fibonacci UA/CG 2584
bases |
|
Marseille4 |
5 |
|
D614G |
6 |
|
UK SN501Y |
6 |
|
Mink |
6 |
|
India B1.617.2 DELTA |
7 |
|
India B.1617 |
7 |
|
SARS-CoV2 Wuhan |
7 |
|
Brazil P1 |
10 |
|
South Afrika B1156 |
12 |
|
South Afrika C1 |
12 |
|
South Afrika B1106 |
12 |
|
South Afrika B1154 |
12 |
|
UK SN510S |
12 |
|
UK SN501T |
12 |
|
Brazil C |
21 |
|
Brazil A |
29 |
|
Brazil B |
34 |
|
Bat RaTG13 |
40 |
|
California CAL20C |
44 |
Table 10: Comparing 19 SARS-CoV2 strains variants « Podium » like left/right Balancing/Unbalancing 1597 UA/CG Fibonacci metastructures.
|
SPIKES |
Left |
Right |
Distance Left-Right |
|
Marseille4 |
18 |
26 |
-8 |
|
Bat RaTG13 |
28 |
31 |
-3 |
|
South Afrika B1156 |
24 |
26 |
-2 |
|
South Afrika C1 |
24 |
26 |
-2 |
|
South Afrika B1106 |
25 |
26 |
-1 |
|
South Afrika B1154 |
25 |
26 |
-1 |
|
UK SN510S |
25 |
26 |
-1 |
|
UK SN501T |
25 |
26 |
-1 |
|
D614G |
29 |
26 |
3 |
|
UK SN501Y |
29 |
26 |
3 |
|
Brazil A |
29 |
26 |
3 |
|
Brazil P1 |
19 |
15 |
4 |
|
Brazil C |
21 |
15 |
6 |
|
India B1.617.2 DELTA |
30 |
24 |
6 |
|
Brazil B |
34 |
26 |
8 |
|
SARS-CoV2 Wuhan |
37 |
26 |
11 |
|
Mink |
41 |
26 |
15 |
|
India B.1617 |
41 |
26 |
15 |
|
California CAL20C |
44 |
26 |
18 |
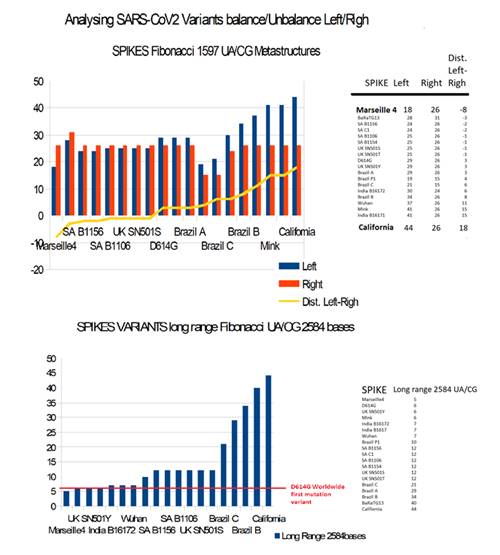
Figure 17: Comparing 19 Spikes from 19 variants for long range 2584 UA/CG and 1597 UA/CG Balancing/Unbalancing « Podium » like metastructures.
Genome analysis:
The few Figures below show how certain genomes of these same 19 variants will, here too, assert - at the scale of the entire genome - the pathogenicity / infectivity of bat RaTG13, Marseille4 or of California Cal.20C, but also of the last Indian B variant B.1.617.2.
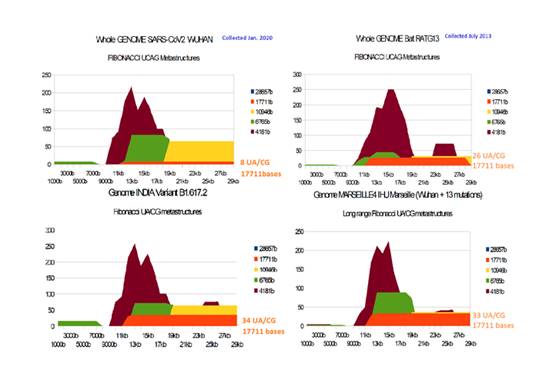
Figure 18: Comparing long range genome overlapping 17711 UA/CG Fibonacci metastructures between SARS-CoV2 reference Wuhan, Bat RaTG13, Marseille4 and India B.1.617.2.
We notice the diversity of spike and genome situations: for Bat RaTG13, the Fibonacci metastructures are very superior to those of the SARS-CoV2 Wuhan simultaneously for spike (5.7 times) and for genome (3.2 times). On the contrary, for Marseille4, only 13 mutations make it possible to multiply by more than 4.2 the Fibonacci of the genome but not of the Spike. Likewise, for India B.1.617.2, there are 23 mutations which lead to a similar situation (4.1 times).
Finally, we could propose a causal link between vaccines and variants as suggested in (Megawaty Tan et al, 2021).
"We can add that the evolution of
the virus towards the" Fibonacci "variants was favored by the
anti-spike protein antibodies of the original SARS-CoV2 virus from Wuhan.
Nature or God will not facilitate the reaction of vaccinators to end to this
pandemic ". Luc Montagnier.
ADE, Variants, Fibonacci "podium
like" unbalancing metastructures and MASTER CODE
SPIKES consistency: is there a possible correlation?
One fact is clear, FIBONACCI structures increase as new variants emerge.
On the one hand, we have just highlighted the possible imbalances of the “podium-like” structures of the very large fibonacci metastructures of the Spikes of the various variants.
We have just classified and sorted these different variants according to these imbalances (Figure 17).
On the other hand, the so-called MASTER CODE theory (Perez, 2018) makes it possible to quantify the quality of the Genomics / Proteomics coupling of any genetic sequence. We had already used this technique in the context of SARS-CoV2: to analyze hypothetical regions of the genome manipulated in the laboratory (Perez§Montagnier, 2020) or, more recently, to highlight the chaotic nature (saturation in CG bases) spikes from Pfizer or Moderna vaccines (Perez, 2021).
We wanted here to search - for the SPIKES of these different variants - for possible CORRELLATIONS between, on the one hand, the Fibonacci imbalances described above, and the respective Genomics / Proteomics couplings of these same SPIKES sequences.
If we discover a possible correlation, it would mean that these variants simultaneously reinforce their mRNA (Fibonacci) structure and the "quality" of the protein produced, which is indeed what the MASTER CODE measures in a way. But in this specific case we do not know a possible link with the pathogenicity of these variants.
Table 11 below presents a rather positive and encouraging result: more than 40% of correlation between these 2 phenomena which appear to be totally independent ....
Table 11: Correllating 17 SARS-CoV2 strains variants « Podium » like left/right Balancing/Unbalancing 1597 UA/CG Fibonacci metastructures with MASTER CODE Genomics/Proteomics % coupling.
|
SPIKES |
Left |
Right |
DIST1 : VARIANTS SPIKES UNBALANCING
Distance Left-Right |
MAST1 : VARIANTS SPIKES MASTER CODE Genomics/Proteomics coupling % |
|
Marseille4 |
18 |
26 |
-8 |
63.01 |
|
South
Afrika B1156 |
24 |
26 |
-2 |
61.98 |
|
South
Afrika C1 |
24 |
26 |
-2 |
61.98 |
|
South
Afrika B1106 |
25 |
26 |
-1 |
62.91 |
|
South
Afrika B1154 |
25 |
26 |
-1 |
62.91 |
|
UK
SN510S |
25 |
26 |
-1 |
62.33 |
|
UK
SN501T |
25 |
26 |
-1 |
63.66 |
|
D614G |
29 |
26 |
3 |
62.91 |
|
UK
SN501Y |
29 |
26 |
3 |
63.85 |
|
Brazil
A |
29 |
26 |
3 |
62.51 |
|
Brazil
P1 |
19 |
15 |
4 |
63.35 |
|
Brazil
C |
21 |
15 |
6 |
65.06 |
|
India B1.617.2 DELTA |
30 |
24 |
6 |
64.42 |
|
Brazil
B |
34 |
26 |
8 |
62.68 |
|
SARS-CoV2
Wuhan |
37 |
26 |
11 |
62 |
|
India B.1617REF |
41 |
26 |
15 |
63.14 |
|
California CAL20C |
44 |
26 |
18 |
64.94 |
|
|
|
|
40.35% correlation |
|
Results of the correlation;
DIST1
¯8 ¯2 ¯2 ¯1 ¯1 ¯1 ¯1 3 3 3 4 6 6 8 11 15 18
MAST1
63.01 61.98 61.98 62.91 62.91 62.33 63.66 62.91 63.85 62.51 63.35 65.06 64.42 62.68 62 63.14 64.94
DIST1 CORRELL MAST1
0.4035350341
then, 40.35% correlation between the 2 phenomena.
These properties brought to light with the variants make it possible to alert more particularly to this risk of “ADE” due to vaccines against SARS-CoV2.
“The paradox is that the vaccinated
people are made more susceptible to infection by variant viruses in epidemic
circulation. It is a well-known ADE effect in RNA viruses which risks being
catastrophic particularly for nursing staff.” Luc Montagnier.
“The paradox is AN OBSERVATION FACT: those vaccinated against the Cov19 spike protein are more infected with the new delta, lambda variants, etc. These variants are the most distant from the SARS-CoV2 Wuhan initial strain so are in the ideal conditions to induce ADEs.” Luc Montagnier.
What is an ADE?
ADE: Facilitating antibodies, ADE (antibody-dependent enhancement) or VAED (vaccine-associated enhanced disease). An ADE stricto sensu where facilitating antigen-antibody complexes bind to the FcgammaRII (CD32) receptors of the membrane of immune cells (mainly monocytes, macrophages and dendritic cells, sometimes B lymphocytes), which promotes their infection by the virus involved.
The existence of facilitating antibodies in COVID-19: In other words, heterotypic antibodies at low levels (resulting from the covid19 vaccine) are responsible for ADE in people infected with a virus serotype (variant) different from the first infection (original sarscov2 strain targeted by the vaccine.).
For the first time the work of JC Perez
allows the detection of numerical series in the natural sequence evolution of
new variants of Covid-19 Corona virus.
Long Fibonacci séries
are described by him in the variants which are the most spreading in the human
population.
This would indicate a natural selection
of more stable structures also possibly more transmissible.
This evolution is in contrast with the
path followed by the vaccine makers:
to make the synonymous codons enriched in G-C in order to increase their m-RNA vaccine stability.
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The author have declared that no competing interests exist.
Thanks for fructuous discussions about this article to Megawaty Tan (A private researcher based in South Sumatera, Indonesia), Alexandra Henrion-Caude ( Future of Research Team, SimplissimA International Research Institute, 39 rue saint Louis, 11324 Port-Louis, Mauritius), Sami MacKenzie-Kerr private researcher in Indonesia ("The Matrix", https://www.google.com/url?sa=t&source=web&rct=j&url=https://matrix.fandom.com/wiki/The_Matrix/Crew&ved=2ahUKEwjf6u7t0NXvAhUBCxoKHRQIA0IQFjAPegQIBxAC&usg=AOvVaw1OcoHfMy2CVksJjUqkvBpU ), Robert Friedman M D. (author of "Nature's secret nutrient, golden ratio biomimicry, for PEAK health, performance and longevity), Philip Risby (initiator of "Learning to Survive") project in Portugal, Valère Lounnas, (Free-lance researcher at CMBI European Molecular Biology Laboratory (EMBL) Heidelberg ), Jacques Demongeot ( Laboratory AGEIS EA 7407, Faculty of Medicine, University of Grenoble Alpes, 38700 La Tronche, France ). Dr Daniel Favre, independant researcher, Brent, Switzerland, David Bensaid M.D Israel ( www.emi-sion.com ), Christian Marc, ( retired, MSEE-Dipl-Eng Physics, MBA (Beta Gamma Sigma, USA), Harvard HBS Alumn, General Director https://www.caravanedelapaix.com/), Ethirajan Govindarajan (adjunct Professor, Department of Cybernetics, School of Computer Science, University of Petroleum and Energy Studies, Dehradun, Uttarakhand, India, Director, PRC Global Technologies Inc., Ontario, Canada, President, Pentagram Research Centre Pvt. Ltd., Hyderabad, India) and Xavier Azalbert, Director FRANCE-SOIR newspaper ( https://www.francesoir.fr/info-en-direct
We thank particularly dr Richard M Fleming PhD, MD, JD (https://www.flemingmethod.com/ and https://www.francesoir.fr/amp/article/videos-les-debriefings/dr-richard-fleming-son-debriefing) for discussions on SARS-CoV2 origins and prion like diseases risk (see https://biomedres.us/fulltexts/BJSTR.MS.ID.000369.php).
Finally, this work is the result of
multiple exchanges and advice, since the very beginning of the COVID-19
pandemic, for which I must thank Professor Luc Montagnier (Nobel
prizewinner for his discovery of HIV, Fondation Luc
Montagnier Quai Gustave-Ador 62 1207 Geneva,
Switzerland).
[1]
(Castro-Chavez,
2020), F. Castro-Chavez, (June 2020), Anticovidian v.2: COVID-19: Hypothesis of the
Lab Origin versus a Zoonotic Event Which Can Also be of a Lab Origin, GJSFR,
August 2020,
https://zenodo.org/record/3988139#.YGMMaq8zaM8
[2]
(Dae Eun Jeong
et al, 2021), Dae Eun Jeong et al,
Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273,
GitHub, March 2021,
https://github.com/NAalytics/Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273
[3]
(Da
Silva Filipe, 2020), da Silva Filipe, A., Shepherd, J.G., Williams, T. et al.
Genomic epidemiology reveals multiple introductions of SARS-CoV-2 from mainland
Europe into Scotland. Nat Microbiol 6, 112–122 (2021). https://doi.org/10.1038/s41564-020-00838-z
[4]
(Demongeot§Henrion-Caude, 2020), Demongeot
J. § Henrion-Caude A., Footprints of a Singular
22-Nucleotide RNA Ring at the Origin of Life, Biology 2020, 9(5), 88;
https://doi.org/10.3390/biology9050088
[5]
(Fournier
et al., 2021) Fournier P.E, Emergence and outcome of
the SARS-CoV-2 “Marseille-4” variant 6 Short title: Outcome of the Marseille-4 genotype,
https://www.mediterranee-infection.com/wp-content/uploads/2020/04/Ms_Marseille-4_IHU-MI_Jan2021vL.pdf
[6]
(Govindarajan,
2020a) Ethirajan Govindarajan et al, “Pairwise
Spatial Correlation of SARS-Corona Viruses”, London Journal of Research in
Computer Science and Technology, London Journals Press, Volume 20, Issue 1,
Compilation 1.0, 2020, pp 11-78
[7]
(Govindarajan,
2020b) Ethirajan Govindarajan et al, “Pairwise
Spectral Correlation of SARS-Corona Viruses”, London Journal of Research in
Computer Science and Technology, London Journals Press, Volume 20, Issue 2,
Compilation 1.0, 2020, pp 81-148
[8]
(Gröhs Ferrareze P. A., et al,
2021), Patrícia Aline Gröhs
Ferrareze, et al, E484K as an innovative phylogenetic
event for viral evolution: Genomic analysis of the E484K spike mutation in
SARS-CoV-2 lineages from Brazil
bioRxiv 2021.01.27.426895; doi:
https://doi.org/10.1101/2021.01.27.426895
[9]
(Hammer
et al, 2021), Hammer AS, Quaade ML, Rasmussen TB, et
al. SARS-CoV-2 Transmission between Mink (Neovison
vison) and Humans, Denmark. Emerg Infect Dis.
2021;27(2):547-551. doi:10.3201/eid2702.203794
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7853580/
[10] (Jackson et al, 2020), Jackson,
N.A.C., Kester, K.E., Casimiro, D. et al. The promise
of mRNA vaccines: a biotech and industrial perspective. npj
Vaccines 5, 11 (2020). https://doi.org/10.1038/s41541-020-0159-8
[11] (Kudla et al, 2016), Kudla, G.,
Lipinski, L., Caffin, F., Helwak,
A. & Zylicz, M. High guanine and cytosine content
increases mRNA levels in mammalian cells. Plos Biol.
4, e180 (2016). High guanine and cytosine content increases mRNA levels in
mammalian cells
[12] (Kustin
T. et al, 2021), Evidence for increased breakthrough rates of SARS-CoV-2
variants of concern in BNT162b2 mRNA vaccinated individuals, Talia Kustin et al, medRxiv Preprints,
Doi: https://doi.org/10.1101/2021.04.06.21254882
[13] (Megawaty
Tan et al, 2021), Megawaty Tan et al. "May
vaccines select SARS-CoV-2 variants more readily escaping immunity – an
analysis of public data". Archives of Microbiology & Immunology, in
press.
[14] (Mengwen
et al, 2006), Mengwen Jia, Liaofu
Luo, The relation between mRNA folding and protein structure,Biochemical and iophysical
Research Communications, Volume 343, Issue 1,2006,Pages 177-182,ISSN 0006-291X,
https://doi.org/10.1016/j.bbrc.2006.02.135. (https://www.sciencedirect.com/science/article/pii/S0006291X06004451)
[15] (Montagnier L. § Kingsley Sanders
F., 1963), Luc Montagnier and F. Kingsley Sanders « Replicative Form of
Encephalomyocarditis virus RNA », Nature 199.
664-667. 1963
[16] (Montagnier L., 2021) video youtube (https://youtu.be/KrIoPIQZmUE)
[17] (Muttineri
et al, 2021), Muttineni R, Kammili
N, Bingi TC, Rao M. R, Putty K, Dholaniya
PS, et al. (2021) Clinical and whole genome characterization of SARS-CoV-2 in
India. PLoS ONE 16 (2): e0246173. doi:
10.1371 / journal. pone.0246173 https://www.google.com/url?sa=t&source=web&rct=j&url=https://journals.plos.org/plospathogens/article/file%3Fid%3D10.1371/journal.pone.0246173%26type%3Dprintable&ved=2ahUKEwj3zdnZnorwAhUQKBoKHUxnD_EQFjABegQICBAC&usg=AOvVaw1A79ux6UbetoPoRx_jT-Mk
[18] (Naveca
Felipe et al, 2021), Phylogenetic relationship of SARS-CoV-2 sequences from
Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y
in the Spike protein, Virological.org, 2021,
https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585
[19] (Perez, 1988), Perez J.C., De nouvelles voies vers l'Intelligence Artificielle, 1988, Ed. MASSON ELSEVIER, EAN 978-2225818158
ISBN 2225818150, https://livre.fnac.com/a223887/Jean-Claude-Perez-De-Nouvelles-voies-vers-l-intelligence-artificielle
[20] (Perez, 1991), J.C. Perez (1991),
"Chaos DNA and Neuro-computers: A Golden Link", in Speculations in
Science and Technologyvol. 14 no. 4, ISSN 0155-7785,
January 1991 Speculations in Science and Cell Motility 14(4):155-7785 https://www.researchgate.net/publication/258439719_JC_Perez_1991_Chaos_DNA_and_Neurocomputers_A_Golden_Link_in_Speculations_in_Science_and_Technologyvol_14_no_4_ISSN_0155-7785
[21] (Perez, 1997), Perez J.C, L'ADN décrypté, Ed. Marco Pietteur,
ISBN: 2-87211-017-8
EAN: 9782872110179,
https://www.editionsmarcopietteur.com/resurgence/91-adn-decrypte-9782872110179.html
[22] (Perez, 2009), Perez J.C, Codex
biogenesis – Les 13 codes de l'ADN (French Edition)
[Jean -Claude ... 2009);
Language: French; ISBN -10: 2874340448; ISBN -13:
978-2874340444
https://www.amazon.fr/Codex-Biogenesis-13-codes-lADN/dp/2874340448
[23] (Perez, 2015), Deciphering Hidden
DNA Meta-Codes -The Great Unification & Master Code of Biology, journal of Glycémies abd Lipidomics,
https://www.longdom.org/abstract/deciphering-hidden-dna-metacodes-the-great-unification-amp-master-code-of-biology-11590.html,
ISSN: 2153-0637, DOI: 10.4172/2153-0637.1000131
[24] (Perez, 2017), J.C Perez, 2017,
Sapiens Mitochondrial DNA Genome Circular Long Range Numerical Meta Structures
are Highly Correlated with Cancers and Genetic Diseases mtDNA
Mutations
January 2017 Journal of Cancer
Science and Therapy 09(06) DOI: 10.4172/1948-5956.1000469
[25] (Perez, 2017b), Jean Claude Perez,
"The Master Code of Biology: Self-assembly of two identical Peptide’s beta
A4 1-43 Amyloid in Alzheimer’s Diseases,"
Biomedical Journal of Scientific & Technical Research, Biomedical Research
Network+, LLC, vol. 1(4), pages 1191-1195, September. Handle: RePEc:abf: journl: v:1: y:2017: i:4:
p:1191-1195 DOI: 10.26717/BJSTR.2017.01.000394
[26] (Perez, 2017c), Perez JC (2017)
The “Master Code of DNA”: Towards the Discovery of the SNPs Function
(Single-Nucleotide Polymorphism). J Clin Epigenet.
3:26. doi: 10.21767/2472-1158.100060,
https://clinical-epigenetics.imedpub.com/the-master-code-of-dna-towards-the-discovery-of-the-snps-function-singlenucleotide-polymorphism.pdf
[27] (Perez, 2018), Perez, J.C. Six
Fractal Codes of Biological Life:perspectives in
Exobiology, Cancers Basic Research and Artificial Intelligence Biomimetism Decisions Making. Preprints 2018, 2018090139 (doi: 10.20944/preprints201809. 0139.v1). Perez, J.C. Six
Fractal Codes of Biological Life:perspectives in
Exobiology, Cancers Basic Research and Artificial Intelligence Biomimetism Decisions Making. Preprints 2018, 2018090139 (doi: 10.20944/preprints201809. 0139.v1).
https://www.preprints.org/manuscript/201809.0139/v1
[28] (Perez, 2019), Perez, J.
Epigenetics Theoretical Limits of Synthetic Genomes: The Cases of Artificials Caulobacter (C. eth-2.0), Mycoplasma Mycoides
(JCVI-Syn 1.0, JCVI-Syn 3.0 and JCVI_3A), E-coli and YEAST chr
XII. Preprints 2019, 2019070120 (doi:
10.20944/preprints201907. 0120.v1).
https://www.preprints.org/manuscript/201907.0120/v1
[29] Perez J.C, (2020). “WUHAN COVID-19
SYNTHETIC ORIGINS AND EVOLUTION.”International
Journal of Research - Granthaalayah, 8(2), 285-324. https://doi.org/10.5281/zenodo.3724003
[30] (Perez§Montagnier,
2020a), Perez, j.c, & Montagnier, L. (2020, April
25). COVID-19, SARS and Bats Coronaviruses Genomes
unexpected Exogeneous RNA Sequences. https://doi.org/10.31219/osf.io/d9e5g
[31] (Perez%Montagnier,
2020b), Jean claude Perez, & Luc Montagnier.
(2020). COVID-19, SARS AND BATS CORONAVIRUSES GENOMES
PECULIAR HOMOLOGOUS RNA SEQUENCES. International Journal of Research
-GRANTHAALAYAH ISSN (print): 2394-3629 July 2020, Vol 8(07), 217 – 263 DOI:
Https://doi.org/10.29121/granthaalayah.v8.i7.2020.678, Vol 8(07), 217 – 263(Vol
8(07), 217 – 263), Vol 8(07), 217–Vol 8(07), 263.
http://doi.org/10.5281/zenodo.3975578
[32] (Perez, 2021) Perez, J. SARS-CoV2
Variants and Vaccines mRNA Spikes Fibonacci Numerical UA/CG Metastructures.
Preprints 2021, 2021040034 (doi: 10.20944/preprints202104.
0034.v5). Perez, J. SARS-CoV2 Variants and Vaccines mRNA Spikes Fibonacci
Numerical UA/CG Metastructures. Preprints 2021,
2021040034 (doi: 10.20944/preprints202104. 0034.v5).
https://www.preprints.org/manuscript/202104.0034/v5
[33] (Pragya Yadav et al., 2021),
Pragya Yadav, et al., Neutralization of variant under investigation B.1.617
with sera of BBV152 vaccinees bioRxiv
2021.04.23.441101; doi:
https://doi.org/10.1101/2021.04.23.441101
[34] (Rapoport § Perez, 2018), Rapoport
D. § Perez J.C, Golden ratio and Klein bottle Logophysics: The Keys of the Codes of Life and Cognition.
Quantum Biosystems. 9(2) 8-76.; Vol. 9 – n.2 – 2018
[35] (Simmonds P, 2020), P. Simmonds,
Rampant C->U hypermutation in the genomes of SARS-CoV-2 and other
coronaviruses – causes and consequences for their short and long evolutionary
trajectories. bioRxiv 2020.05.01.072330; doi: https://doi.org/10.1101/2020.05.01.072330
[36] (Srivastava Surabhi et al, 2021),
SARS-CoV-2 genomics: An Indian perspective on sequencing viral variants, J Biosci. 2021; 46(1): 22. Published online 2021 Feb 20. doi: 10.1007/s12038-021-00145-7,
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7895735/
[37] (Wenjuan
Zhang et al, 2021), Emergence of a novel SARS-CoV-2 strain in Southern
California, USA, medRxiv 2021.01.18.21249786; doi: https://doi.org/10.1101/2021.01.18.21249786.
 This work is licensed under a: Creative Commons Attribution 4.0 International License
This work is licensed under a: Creative Commons Attribution 4.0 International License
© Granthaalayah 2014-2021. All Rights Reserved.

