
|
|
|
|
COVID-19, SARS AND BATS CORONAVIRUSES GENOMES PECULIAR HOMOLOGOUS RNA SEQUENCESJean Claude Perez *1
|
|
|
|


Article Type: Research Article
Article Citation: Perez, J. C. Montagnier, L.. (2020). COVID-19,
SARS AND BATS CORONAVIRUSES GENOMES PECULIAR HOMOLOGOUS RNA SEQUENCES. International
Journal of Research -GRANTHAALAYAH, 8(7), 217-263. https://doi.org/10.29121/granthaalayah.v8.i7.2020.678
Received Date: 07 July 2020
Accepted Date: 30 July 2020
Keywords:
COVID-19
Bats Coronaviruses
RNA Sequences
SARS
HIV
Plasmodium yoelii
Spike
ABSTRACT
We are facing the worldwide invasion of a new coronavirus. This follows several limited outbreaks of related viruses in various locations in a recent past (SARS, MERS). Although the main current objective of researchers is to bring efficient therapeutic and preventive solutions to the global population, we need also to better understand the origin of the newly coronavirus-induced epidemic in order to avoid future outbreaks. The present molecular appraisal is to study by a bio-infomatic approach the facts relating to the virus and its precursors.
This article shows how 16 fragments (Env Pol and Integrase genes) from different strains, both diversified and very recent, of the HIV1, HIV2 and SIV retroviruses have high percentage of homology into parts of the genome of COVID_19. Moreover each of these elements is made of 18 or more nucleotides and therefore may have a function. They are called Exogenous Informative Elements (EIE).
Among these EIE, 12 are concentrated in a very small region of the COVID-19 genome, length less than 900 bases, i.e. less than 3% of the total length of this genome. In addition, these EIE are positioned in two functional genes of COVID-19: the orf1ab and S spike genes.
Here are the two main facts which contribute to our hypothesis of a partially synthetic genome: A contiguous region representing 2.49% of the whole COVID-19 genome of which 40.99% is made up of 12 diverse fragments originating from various strains of HIV SIV retroviruses. Some of these 12 EIE appear concatenated. Notably, the retroviral part of these regions, which consists of 8 elements from various strains of HIV1, HIV2 and SIV covers a length of 275 contiguous bases of COVID-19. The cumulative length of these 8 HIV/SIV elements represents 200 bases. Consequently, the HIV SIV density rate of this region of COVID-19 is 200/275 = 72.73%.
A major part of these 16 EIE already existed in the first SARS genomes as early as 2003. However, we demonstrate how a new region including 4 HIV1 HIV2 Exogenous Informative Elements radically distinguishes all COVID-19 strains from all SARS and Bat strains with the exception of Bat RaTG13.
We gather facts about the possible origins of COVID_19. We have particularly analyzed this small region of 225 bases common to COVID_19 and bat RaTG13.
We have studied the most recent genetic evolution of the COVID_19 strains involved in the world epidemic. We found a significant occurrence of mutations and deletions in the 225 bases area.
On sampling genomes, we show that this 225 bases key region of each genome, rich in EIE, and the 1770bases SPIKE region evolve much faster than the corresponding whole genome (cases of 44 patients genomes from WA Seattle state, original epicenter in USA).
In the comparative analysis of both SPIKES genes of COVID_19 and Bat RaTG13 we note two abnormal facts:
1) the insertion of 4 contiguous PRRA amino acids in the middle of SPIKE (we show that this site was already an optimal cleavage site BEFORE this insertion).
2) an abnormal distribution of synonymous codons in the second half of SPIKE.
Finally we show the insertion in this 1770 bases SPIKE region of a significant pair of EIEs from Plasmodium Yoelii and of apossible HIV1 EIE with a crucial Spike mutation.
1. INTRODUCTION
We are facing the worldwide invasion of a new coronavirus. This follows several limited outbreaks of related viruses in various locations in a recent past (SARS, MERS) [1], [2]. The human civilization has been very successful in the last centuries regarding demographic and economic growths. However, in our times, the economic power is concentrated in the hands of a few individuals and consequently economic interests are prevailing over the well being of humanity.
Although the main objective of researchers is to bring efficient therapeutic and preventive solutions to the global population, we also need to better understand the origin of the new coronavirus-induced epidemic in order to avoid future outbreaks. The present molecular appraisal is to study by a bio-infomatic approach the facts relating to the virus and its precursors.
We had analyzed the evolution of coronaviruses from the first SARS (2003), to the first genomes of COVID- 19, when it was still called 2019-nCoV [3]. We had knowledge of the online article by J.Lyons-Weiler [4] according to which a region of around 1kb is totally new in the genome of COVID-19.
Using our proprietary bio-mathematic approach where we are able to evaluate the level of cohesion and organization of a genome, we discovered that the deletion by mutation of this new region of 1kb [4] would increase the level of «structural harmonization» of the genome.
This suggests a possible exogenous «addition» to the genome. Upon studying the publication of Pradhan et al. [15] we then searched in this genome for possible traces of HIV or even SIV. A first publication [5] reports the discovery of 6 HIV SIV RNA pieces relates to crucial retroviral genes like Envelope and RT Pol. The present article confirms and extends these initial results.
2. MATERIALS AND METHODS
2.1. ACCESS TO DATA BANKS
Preliminary Note
The COVID-19 genome sequence initially studied for this article is NC_045512.2. More generally, we are interested in the first genomes published under the reference "Wuhan market". However, these sequences published in January 2020 evolved somewhat during the first quarter of 2020. Thus, NC_045512.2 has evolved from 29866 bases to 29903 bases; so, our Genbank NCBI reference was also changed.
All these sequences of genomes referenced as "Wuhan market" relating to individual patients, were deposited on January 30, 2020 and then re-published on March 6, 2020. For these reasons we will have to specify and adjust here the addresses of the key regions "A" and "B " which we analyze in this article.
The Wuhan market referenced genomes are presently:
https://www.ncbi.nlm.nih.gov/nuccore/LR757995.1
https://www.ncbi.nlm.nih.gov/nuccore/LR757996.1
https://www.ncbi.nlm.nih.gov/nuccore/LR757997.1
https://www.ncbi.nlm.nih.gov/nuccore/LR757998.1
and
https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2
Thus, the start address of the region of 330 bases named in this article "region B" which was initially positioned at 21673 bases in our previous article is now shifted at 21698 bases in NC_045512.2 , at 21683b in LR757995.1, at 21678 bases in LR757996.1, , and at 21673 bases in LR757998.1. The sequence LR757997.1, is unavailable because it contains more than 10,000 indeterminate « N » bases.
Finally, this region « B » has the same starting address in our NC_045512.2 reference sequence and in LR757998.1. The reference sequence used in this article is: https://www.ncbi.nlm.nih.gov/nuccore/LR757998.1
So, we use as reference the former referenced genome: Wuhan market ID: LR757998.1
Validation of nucleotide fragments as «Exogenous
Informative Elements» (EIE):
We have chosen this minimal length of 18 nucleotides (6 amino acids) for the support of information (thus as an antigenic motif). This is also the size of the primers used for PCR which allowing a high specificity of sequence selection on DNA recognition.
Main COVID_19 genes involved
The two main genes involved in COVID-19 genome are Orf1ab and «S» Spike. Their relative addresses in our referenced genome are:
266... 21555 for Orf1ab
21563...25384 for S spike
The main analyzed regions
Region « A », Location of the 600 bases from the COVID_19 reference genome “Wuhan market”
ID: LR757998.1.
Its length was between 21072 and 21672 nucleotides.
AGGGTTTTTTCACTTACATTTGTGGGTTTATACAACAAAAGCTAGCTCTTGGAGGTTCCGTGGCTATAAAGATAACAGAACATTCTTGGAATGCTGATCTTTATAAGCTCATGGGACACTTCGCATGGTGGACAGCCTTTGTTACTAATGTGAATGCGTCATCATCTGAAGCATTTTTAATTGGATGTAATTATCTTGGCAAACCACGCGAACAAATAGATGGTTATGTCATGCATGCAAATTACATATTTTGGAGGAATACAAATCCAATTCAGTTGTCTTCCTATTCTTTATTTGACATGAGTAAATTTCCCCTTAAATTAAGGGGTACTGCTGTTATGTCTTTAAAAGAAGGTCAAATCAATGATATGATTTTATCTCTTCTTAGTAAAGGTAGACTTATAATTAGAGAAAACAACAGAGTTGTTATTTCTAGTGATGTTCTTGTTAACAACTAAACGAACAATGTTTGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTTAATCTTACAACCAGAACTCAATTACCCCCTGCATACACTAATTCTTTCACACGTGGTGTTTATTACCCTGACAAAGTTTTCAGATCC
See details alignment in supplementary materials « a ».
Region «B», Location of the 330 first bases from the COVID_19 reference genome “Wuhan market”
ID: LR757998.1.
Their length was between 21672 and 22002 nucleotides (then immediately following region «A»:
TCAGTTTTACATTCAACTCAGGACTTGTTCTTACCTTTCTTTTCCAATGTTACTTGGTTCCATGCTATACATGTCTCTGGGACCAATGGTACTAAGAGGTTTGATAACCCTGTCCTACCATTTAATGATGGTGTTTATTTTGCTTCCACTGAGAAGTCTAACATAATAAGAGGCTGGATTTTTGGTACTACTTTAGATTCGAAGACCCAGTCCCTACTTATTGTTAATAACGCTACTAATGTTGTTATTAAAGTCTGTGAATTTCAATTTTGTAATGATCCATTTTTGGGTGTTTATTACCACAAAAACAACAAAAGTTGGATGGAAAGT
See details alignment in supplementary materials « b ».
We analyzed this larger region which starts at the same address as our region "B":
entitled « Region Lyons-Weiler » [4].
Their length was between 21672 and 23050 (1378 nucleotides) within reference genome Wuhan market
ID: LR757998.1
In the RESULTS and DISCUSSION, we will more particularly analyze a small region of 225 nucleotides of the reference genome:
TGTTTTTCTTGTTTTATTGCCACTAGTCTCTAGTCAGTGTGTTAATCTTACAACCAGAACTCAATTACCCCCTGCATACACTAATTCTTTCACACGTGGTGTTTATTACCCTGACAAAGTTTTCAGATCCTCAGTTTTACATTCAACTCAGGACTTGTTCTTACCTTTCTTTTCCAATGTT ACTTGGTTCCATGCTATACATGTCTCTGGGACCAATGGTACTAA
Alignments: Analyzing COVID-19 DNA sequences, We use BLAST NCBI (National Center for Biotechnology) public tool.
BLASTn - NIH
NCBI National Center for Biotechnology Information.
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch
Relating the « DNA Master Code », a biomathematic method
to analyze cohesion/heterogeneity of a DNA/RNA sequence:
We must introduce and summarize this theoretical method, because it constitutes a strong way to illustrate crucial differences between COVID_19 and bat RaTG13 specific genomes (Figs 4, 5, 12 and 13).
Full details on this numerical method in [6], [7], [8], and [31], and recall Methods in supplementary Materials « 9 »..
Starting from the atomic masses of the C O N H S P bioatoms constituting RNA, DNA nucleotides and amino acid, a simple law of projection of these atomic masses leads to a UNIFICATION of GENOMICS and PROTEOMICS patterned images that can be calculated for any DNA/RNA codons sequence. This numerical projection of atomic masses produces a whole numbers numerical code common to the triplets codons DNA, RNA, or amino acids. A process of DIGITAL INTEGRATION at short, medium and very long distance then allows a globalization of genetic information by a principle which recalls an analogy with the HOLOGRAM.
« Thus, any codon radiates at long distance and vice versa ». The Master Code of this sequence then produces two signatures, one GENOMIC and the other for PROTEOMIC, materialized by 2 very strongly correlated curves. And is this level of coupling which will provide key information on the COHESION or on the HETEROGENEITY [11] of this nucleotide sequence. in particular the extreme regions (mini / maxi) would be associated with biological functions such as active sites, chromosomes breakpoints, etc.
Dynamics of the COVID_19 sequences available:
We will specify that this study having been carried out over several weeks at the time when the number of genomes of COVID_19 was constantly evolving, we saw fit to specify, each time in deital characters, the dates of the BLASTn searches as well as the number of sequences available at this exact moment.
3. RESULTS AND DISCUSSION
This RESULTS and DISCUSSION will have 4 main sections:
Part I
18 RNA fragments of homology equal or more than 80% with human or simian retroviruses have been found in the COVID-19 genome. These fragments are 18 to 30 nucleotides long and therefore have the potential to modify the gene expression of Covid19. We have named them Exogenous Informative Elements or EIE. These EIE are not dispersed randomly, but are concentrated in a small part of the genome (§1 and 2).
Part II
This region, a 225-nucleotide long region is unique to COVID_19 and Bat RaTG13 and can also discriminate between these 2 genomes (§3, 4, 5 6 and 7).
Part III
In the decreasing slope of the epidemic, this 225 bases area exhibits an abnormally high rate of mutations/deletions, particularly in the USA Seattle WA state (§8, 9 and 10).
Part IV
The comparative analysis of the SPIKES genes of COVID_19 and of Bat RaTG13 (§11, 12, 13 and 14).
Part I
18 RNA fragments of homology equal or more than 80% with human or simian retroviruses have been found in the COVID_19 genome. These fragments are 18 to 30 nucleotides long and therefore have the potential to modify the gene expression of Covid-19. We have named them Exogenous Informative Elements or EIE. These EIE are not dispersed randomly, but are concentrated in a small part of the genome (§1 and 2).
Warning: on the limits of bioinformatics tools like BLASTn: the main criticism that this article will have to face is that of the relevance of our BLASTn analyzes highlighting many small traces of HIV in the genome of COVID_19. We will answer with the following 2 facts:
1) We limit the HIV fragments selected to a minimum of 18 bases to consider them as relevant.
2) Today, technologies such as CRISPR-Cas13 RNA [23] make it possible to modify RNA sequences with a clockmaker's precision capable of placing exogenous sequence fragments "side by side", as we will demonstrate here.
1. A
high density of HIV SIV regions that are diverse both in their nature and in
their collection dates: indeed, a concentration of 12 significant HIV SIV EIE
in only 744bases.
We are looking here for possible traces of HIV1, HIV2 or SIV EIE into our Wuhan market reference genome
LR757998.1.
We will only use as significant EIE those which have at least 18 nucleotides of homology, i.e. 6 codons.
Note: We will present below 12 +4 HIV/SIV EIE in the sequential order of their locations within COVID_19 genome. Initially, by focusing on the genome region mentioned in [4], we find and published [5] 6 first EIE located at the very beginning of this region.
By amore in-depth exploration of this region (region "B" 330 bases), then exploring region "A"
(of 600 bases) immediately located upstream of this region "B ", we discover, concentrated on less than 930 bases, 12 HIV SIV EIE. We complete them with the last 4 EIE located upstream in the genome. It is this set of 16 EIE which will be detailed below.
Evidence for 12 HIV/SIV EIE sequences in regions “A” and
“B” of the COVID-19 genome (plus two in the interface space, one merged and one
overlapped):
Following, the 14 HIV/SIV “Exogenous Informative Elements”:
==> ==> BLASTn detailed scans are in Supplementary Materials (Ref1).
Region A: 600 bases (21072 to 21672)
Details:
Hiv-2. France (2012) 66-81
Hiv-1 Sweden (2017) 154-174
Hiv-2 Guinea (2012) 236-253
SIV Africa (2016) 366-386
Interface:
HIV-1 Kenya (2008) 471-501
HIV-1 Cape Verde (2012) 512-529
Region B: 330 bases (21672 to 22002)
Details:
Hiv-2. Côté ivoire (2014) 23 42 *
Siv Tanzania (2016) 29 50 partial overlap
Siv P18 Africa (2016) 77 96 *
Hiv-1. Netherlands (2016). 85. 112. Usa (2011) 85 108 (merged) *
Hiv-2 UC1 Cote d'Ivoire (1993) 132 157 *
Hiv-2 Sénégal (2011) 179 194 *
Hiv-1 Malawi (2013) 212 243 *
Hiv-1. Russia (2010) 242 280 *
SivagmTan-Cameroon (2015) 279 298 *
We consider only the 8 (*) HIV SIV motifs, the 9th is partially in overlap.
These 14 HIV/SIV -EIE- are detailed in SUPPLEMENTARY MATERIALS (ref 1). They are summarized in Table1.
Table 1: Synoptic table of 12 significant EIE from HIV SIV strains in the "A" and "B" regions of the COVID-19 genome (plus two in the interface).
|
Origins |
HIV
SIV type |
Relative
Location |
«
Exogenous Informative Element » Label |
Genba
nk Access |
Homology |
Bases
identities |
O R F
1 a b |
S s p i
k e |
Real
location |
|
|
Region
A: 600 bases: 21072 to 21672 |
||||||||||
|
266.
21555. Orf1ab. Relative locations 484/600 (end Orf1ab gene), |
||||||||||
|
2012 France |
HIV2 |
66-81 |
HIV-2 isolate 56 from
France envelope glycoprotein (env) gene, partial cds |
JN230
738.1 |
100,00% Unsignific
ant |
16/16 Unsignif
icant |
§ |
|
21137 21152 |
|
|
2017 Sweden |
HIV1 |
154-174 |
3.1 |
100,00% |
21/21 |
§ |
|
21225 21245 |
||
|
2012 Guinea |
HIV2 |
236-253 |
HIV-2
isolate CA65410.13 from Guinea-Bissau envelope gene, partial cds |
3831. |
94,00% |
17/18 |
§ |
|
21307 21324 |
|
|
|
|
|
|
1 |
|
|
|
|
||
|
2016 Africa |
SIV |
366-386 |
Simian immunodeficiency
virus isolate VSAA2001, complete genome |
1 |
95,00% |
20/21 |
§ |
|
21437 21457 |
|
|
21563..25384. |
S
spike |
|
|
|
|
|
|
|||
|
2008 Kenia
[9] |
HIV1 |
471-501 |
HIV-1
clone ML1592n
from Kenya nonfunctional vpu protein (vpu) gene, complete sequence; and
nonfunctional envelope glycoprotein (env) gene,
partial sequence |
1 |
88,00% |
28/32 |
§ |
§ |
21542 21572 |
|
|
2012
Cap verde |
HIV2 |
512-529 |
HIV-2
isolate 05HANCV37 from Cape Verde envelope glycoprotein (env) gene, partial
cds |
7434. |
100,00% |
18/18 |
|
§ |
21583 21600 |
|
|
|
|
|
|
1 |
|
|
|
|
||
|
Region
B: 330 bases (21672 to 22002) |
||||||||||
|
2014 Cote
d'ivoire |
HIV2 |
23-42 |
HIV-2 isolate 106CP_RT from Cote d'Ivoire reverse transcriptase gene, partial cds |
1 |
95,00% |
19/20 |
|
§ |
21694 21713 |
|
|
2016 |
SIV |
29-50 |
Simian
immunodeficiency virus |
1 |
91,00% |
20/22 |
|
§ |
21700 |
|
|
Tanzania |
|
|
isolate
TAN5 from Tanzania, |
|
|
|
21721 |
|||
|
Partially |
|
|
complete
genome |
|
|
|
|
|||
|
overlap |
|
|
|
|
|
|
|
|||
Note: « § » indicates location of each HIV / SIV EIE within COVID_19 genome (gene identification). First, it is important to note that all the regions found here are included in one of the 2 main genes of
Evidence for 4 other HIV/SIV EIE sequences in others areas
of COVID-19 genome:
We also found 4 other non-contiguous HIV SIV regions summarized in Table 2 below. Details of these searches in the supplementary materials "d".
==> ==> These 4 HIV/SIV -EIE- are detailed in SUPPLEMENTARY MATERIALS (ref 2). They are summarized in Table 2.
Table 2: Synoptic table of 4 gene EIE motifs from HIV SIV strains in others areas than the "A" and "B" regions of the COVID-19 genome.
Note: « § » indicates location of each HIV / SIV EIE within COVID_19 genome (gene identification).
Table 3: The 17 HIV/SIV EIE according to their homologies with COVID-19 sorted by decreasing % (the merged one from USA is excluded).
|
HIV SIV strain |
COVID-19 gene |
Homology |
|
HIV2 Env France 2012 (non-significant) |
Orf1ab |
100,00% |
|
HIV1 Sweden 2017 (recombinant form in Sweden) |
Orf1ab |
100,00% |
|
HIV2 Env Cape Verde 2012 |
S spike |
100,00% |
|
HIV2 Pol 2011 Senegal (non-significant) |
S spike |
100,00% |
|
SIV Pol 2015 Germany |
Orf1ab |
100,00% |
|
SIV 2016 African Monkey |
Orf1ab |
95,00% |
|
HIV2 RT Pol 2014 Cote d'ivoire |
S spike |
95,00% |
|
SIV Env 2016 Africa |
S spike |
95,00% |
|
HIV2Env 2012 Guinea |
Orf1ab |
94,00% |
|
HIV1 Integrase 2004 USA |
Orf1ab |
93,00% |
|
HIV1 Env 2011 USA |
Orf1ab |
93,00% |
|
SIV 2016 Tanzania |
S spike |
91.00% |
|
HIV1 Env 2016 Netherlands |
S spike |
89,00% |
|
HIV1 Env 2008 Kenia |
Orf1ab and S spike |
88,00% |
|
HIV1 Env 2013 Malawi |
S spike |
88,00% |
|
HIV1 Env 2016 China |
Orf1ab |
87,00% |
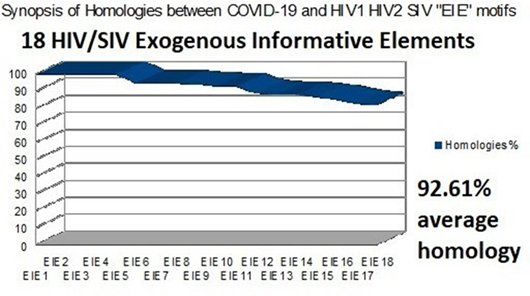
Figure 1: The 18 HIV SIV EIE according to their homologies with COVID-19 sorted by decreasing %.
First, it is important to note that all the regions found here are included in one of the two main genes of COVID-19, so they are «Informative Exogenous Elements». A synthetic chart is in Fig 1.
Some significant results relating to this analyzed region of 930 base pairs (600 + 330) are:
The entire genome has 29903 bases. At least 12 regions are located between the bases 21225 and 21969, which is exactly 744bases.
This therefore represents an average space of 744/12 = 62 bases for each EIE. Or as a % of the whole genome 744/29903 = 2.49% of the whole genome.
As the cumulative length of the 12 EIE is 305 bases, we deduce that the average size of an insert is 337/12
= 25.4bases.
Finally, we deduce an occupancy rate of the 744bases space by EIE from HIV SIV of 25.4/62 = 40.99%. This percentage is considerable.
So, to summarize: a contiguous region representing 2.49% of the whole COVID-19 genome is 40.99% made up of 12 diverse EIE originating from various strains of HIV SIV retroviruses.
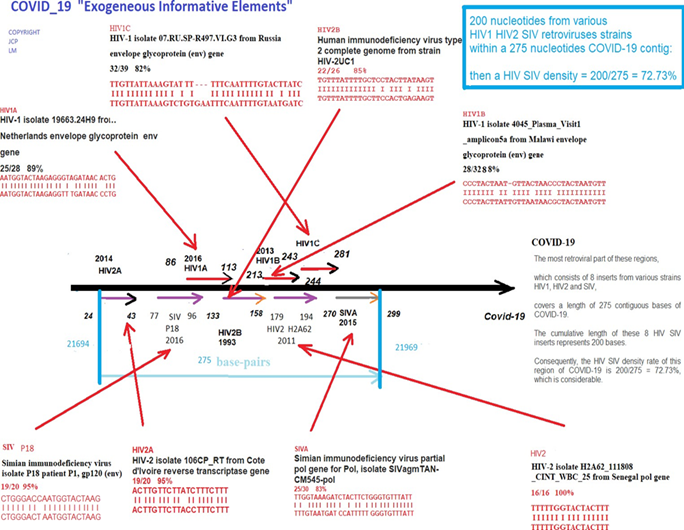
Figure 2: Summary chart of the 8 HIV/SIV EIE from region “B”. This summary chart demonstrating how 200bases from various HIV SIV retroviral strains within a concentrated 275bases COVID-19 contig have a density rate equal to 72.73%.
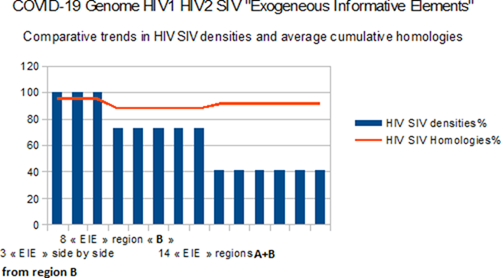
Figure 3: Comparative trends in HIV/SIV EIE densities and average cumulative homologies for 3 clusters.
In these comparative trends in HIV/ SIV EIE densities (blue) and average cumulative homologies (red) for 3 clusters, where 3 region B EIE are side by side, joined by 5 more to complete 8 EIE from region B, plus the final six to integrate all the 14 EIE (A+B cumulated regions).
2. Concatenations
of HIV/SIV regions "placed" in sequence and side by side.
Table 2 shows that two very different EIE follow each other side by side in the RNA sequence of COVID-19:
The first, at location 20373 to 20401 comes from an HIV1 Integrase from a USA virus from 2004 ( Homo sapiens clone HIV1-H9-106 HIV-1 integration site, AY516986.1 ), while the second, at location 20400 to 20430 comes from an Envelope from another HIV1 virus from the USA from 2011 ( HIV-1 isolate JACH1853_A5 from USA envelope glycoprotein (env) gene, complete cds, HQ217329.1 ).
Even more surprisingly, in Table 1, we note the same phenomenon between, this time not 2 but 3 EIE from the radically different HIV SIV viruses:
Here are these 3 EIE concatenated with seemingly perfect " watchmaker's precision":
Malawi, year 2013.
HIV1 212-243 HIV-1 isolate
4045_Plasma_Visit1_amplicon9 Malawi envelope glycoprotein (approx) 88.00% 28/32 Location: 21883 21914
Russia, year 2010.
HIV1 242-280 HIV-1 isolate 07. RU.SP-R497.VI.F5 envelope glycoprotein Russia (env) gene 82.00% 32/39 Location: 21913 21951
Cameroon year 2015.
SIV 279-298 partial simian immunodeficiency virus pol gene for Pol, 83.00% 25/30 Location: 21950 21969
It will be observed that the cumulative length in COVID_19 of these 3 EIE is 126 bases of which the HIV occupied bases are 120. So, a total HIV/COVID_19 of 120/126 > 95%, which is artificially remarkable.
Part II
Within this part, a
225-nucleotide long region is unique to COVID_19 and Bat
RaTG13, and can also discriminate between these 2 genomes (§3, 4, 5, 6 and 7).
The origin of COVID-19 remains an open question: see particularly [14-20] and [5, 27,30, 33, 34].
In this second part of the RESULTS and DISCUSSION, we will present two types of facts: On the one hand, we will show that the 2 genomes of COVID_19 and Bat RaTG13 are exclusively distinguished from all the other genomes of SARS, MERS and other Bats.
On the other hand, we will analyze several specific facts suggesting that COVID_19 does not originate from Bat RaTG13.
3. Evidence
of the absence of 4 HIV/SIV « Exogenous Informative Elements » from COVID_19
within the SARS-2005 and MERS genomes.
In the following Table 4 it appears that 14 of the 18 HIV/SIV EIE existed - already - from the first human SARS genomes that appeared in China around 2003.
However, a novel long region of around 225 nucleotides, less than 1% of the genome, appears to us to have been inserted: This region is completely absent in all SARS genomes, whereas it is present and 100% homologous for all COVID-19 genomes listed in NCBI.
Table 4: Comparing 16 EIE from « A », « B » and remaining regions in COVID-19, HIV/SIV and SARS.
|
HIV/SIV «Exogenous Informative Elements (EIE) » |
Locations within regions of: «A» 600 bases and «B» 330 bases |
Length nucleotides in COVID_19 |
Length nucleotides in HIV and SIV EIE % HIV and SIV / COVID-19 |
Length nucleotides in SARS genomes % SARS/COVID-19 |
|
|
Region « A » |
|||||
|
HIV2 2012 France |
66-81 |
16 non-significant |
16 100% |
13 |
81% |
|
HIV1 2017 Sweden |
154-174 |
21 |
21 100% |
19 |
90% |
|
HIV2 2012 Guinea |
236-253 |
18 |
17 94% |
11 |
61% |
|
SIV 2016 Africa |
366-386 |
21 |
20 95% |
18 |
86% |
|
Start 225 bases zone including 4 «
Exogenous Informative Elements » |
|||||
|
HIV1 2008 Kenia |
471-501 |
32 |
28 88% |
0 |
0% |
|
HIV2 2012 Cap verde |
512-529 |
18 |
18 100% |
0 |
0% |
|
Region « B » |
|||||
|
HIV2 2014 Cote d'ivoire |
23-42 |
20 |
19 95% |
0 |
0% |
|
SIV 2016 Africa |
77-96 |
20 |
19 95% |
0 |
0% |
|
End 225 bases EIE zone including 4 «
Exogenous Informative Elements » (note1) |
|||||
|
HIV1 2016 Netherlands variant HIV1 USA 2011 |
85-112 85-108 |
28 |
25 89% |
13 9 |
46% 32% |
|
HIV2 1993 côte ivoire |
132-157 |
26 |
22 85% |
20 |
77% |
|
HIV2 2011 Sénégal |
179-194 |
16 non-significant |
16 100% |
12 |
75% |
|
HIV1 2013 Malawi |
212-243 |
32 |
28 88% |
22 |
69% |
|
HIV1 2010 russia |
242-280 |
39 |
32 82% |
15 |
38% |
|
SIV 2015 Cameroun. |
279-298 |
30 |
25 83% |
10 |
33% |
|
others areas than the "A" and
"B" regions |
|||||
|
SIV 2015 Germany |
8751 8770 |
20 |
20 100% |
9 |
45% |
|
HIV1 2016 China |
14340 14378 |
38 |
33 87% |
34 |
89% |
|
HIV1 2004 USA |
20373 20401 |
28 |
26 93% |
28 |
100% |
|
HIV1 2011 USA |
20400 20430 |
30 |
28 93% |
21 |
70% |
Note1: this genome HIV-1 USA 2011 is self-contained within the HIV-1 2016 Netherlands variant in the 225 bases area (85-108 and 85-112), the 225 bases frontier is in the relative region “B”.
Here we wanted to find out if the 16 EIE discovered in the COVID-19 genome already existed in the human SARS genomes that appeared in 2003.
Table 4 summarizes this research. In particular, it appears that 14 of the 18 HIV/SIV EIE already existed since the first human SARS genomes that appeared in China around 2003.
However, a novel long region of around 225 nucleotides, appears to us to be totally new: This region is completely absent in ALL SARS genomes, whereas it is present and 100% homologous for all COVID-19 genomes listed in NCBI or GISAID COVID_19 genomic databases.
This region is located (in the COVID-19 genome which served as a reference) between the addresses 21550 and 21772. It is therefore located between the end of region "A" (from base 475 to 600) and the start of region "B" (from base 1 to 99).
A remarkable fact is also observed: the HIV/SIV EIEs which already existed in SARS have evolved a lot through numerous mutations. Thus, four EIEs have very weak homologies (near 30%) between their SARS version and their COVID-19 version. These homologies gradually improve in more recent SARS (2015 or 2017 for example, right column in Table 4).
The 4 « Exogenous Informative Elements » added in COVID_19 are respectively:
HIV1 Kenia 2008
HIV2 Cape Verde 2012
HIV2 Ivory Coast 2014
SIV Africa 2016.
The reader will be able to note that these strains HIV1/HIV2/SIV are very recent and subsequent to the emergence of SARS. However, most of the other strains HIV/SIV (HIV1 2017 Sweden, HIV2 2012 Guinea, etc.) have dates posterior to the emergence of the first SARS. This fact will have to be explained …
The case of the MERS genome:
An analysis of the reference genome of the pathogenic RNA virus MERS ( Middle East respiratory syndrome coronavirus, complete genome NCBI Reference Sequence: NC_019843.3, https://www.ncbi.nlm.nih.gov/nuccore/NC_019843.3?report=genbank ) shows that from the end of our "A" region, and from all of the key 225 base regions, of the "B" region and of the "Lyons-Weiler" region. FOUR crucial regions of our article are totally ABSENT in MERS.
4. Evidence
for HIV/SIV sequences in this region, and their compaction in the 225 bases
portion of both COVID_19 and Bat coronavirus RaTG13 genomes.
We now analyze the level of homologies between the four strains HIV/SIV of the 4 cases which are always present in COVID-19 but always absent in SARS. The remarkable point is as follows: It is strange that the most significant "Bat" genome, Bat coronavirus RaTG13 genome [12], is from 2020, just like COVID-19 ... In particular, for the HIV1 Kenia 2008 sequence [9], [10] bat RaTG13 is the only strain found in the "Bat" population to have it, while for the three other EIE, the "Bat" strains are very numerous but with non-significant HIV/SIV homologies.
Table 5: Comparing the 4 EIE from COVID-19, HIV/SIV and Bat coronavirus RaTG13 [12].
|
HIV/SIV « Exogenous
Informative Elements » |
Locations within
regions of: « A » 600bases and « B
» 330bases |
Length nucleotides in
COVID_19 |
Length nucleotides in
HIV/SIV EIE % HIV-SIV / COVID_19 |
Length nucleotides in Bat
coronavirus RaTG13 genome |
||||
|
Region « A » |
||||||||
|
2008 Kenia HIV1 |
471-501 |
32 |
28 88% |
27 (note1) |
84% |
|||
|
2012 Cap verde HIV2 |
512-529 |
18 |
18 |
100,00% |
16 89% |
(note2) |
||
|
Region « B » |
||||||||
|
2014 HIV2 |
Cote |
d'ivoire |
23-42 |
20 |
19 |
95% |
15 79% |
(note3) |
Note1
COVID-19 / HIV-1 28/32 88%,
Only COVID_19 strains, Bat coronavirus RaTG13 and Rhinolophus affinis
coronavirus isolate LYRa3 spike protein gene. No others Bat strains.
Note2
COVID-19 / HIV-2 18/18 100%, Bat. 16/18. 89%, Sars urbani. 10/10
Various others Bat and Sars with VERY low homologies but all < 10
Note3
COVID-19 / HIV-2 19/20 95%, had a Bat RaTG13. 15/17. 88%. well. Sars urbani. 9/9 Various others Bat and sArs but all <12
Note4
COVID-19 / SIV. 19/20. 95%, Bat coronavirus 10/10, to exchange RNA with bat RaTG13 HIV, Bat. Bad homology. Various Bat and Sars all <12
We must explain why, for HIV1 Kenya, homologies are the
same between COVID_19 and Bat RaTG13, in contrast to the 3 others (Cap verde,
Cote d'ivoire, Africa) where the Bat RaTG13 homologies are lower than those of
COVID_19.
Zooming on the first HIV1 Kenia Homologies:
Synthesis data: Comparing the 3 key regions « A », « B », and « Lyons-Weiler » region [4] in the cases of COVID-19, Bat RaTG13 coronavirus [12] and the best homologies for other Bat and SARS coronaviruses.
Table 6: Comparing the 3 key regions « A », « B », and « Lyons-Weiler » region [4] in the cases of COVID-19, Bat RaTG13 coronavirus [12] and the best homologies for other Bat and SARS coronaviruses.
|
Coronavirus genome |
Region « A » |
Region « B » |
Region « Lyons-weiler » |
|
COVID_19 |
600/600 100% |
330/330 100% |
1378/1378 100% |
|
Bat RaTG13 |
563/599 98% |
309/330 94% |
1209/1311 92% |
|
Other Bat |
518/605 86% (note1a) |
158/212 75% (Note1b) |
402/521 77% (Note1c) |
|
Other SARS |
400/474 84% (note2a) |
144/177 73% (Note 2b) |
297/376 79% (Note2c) |
Note1a - Bat SARS-like coronavirus isolate bat-SL-CoVZC45
Note1b - BtRs-BetaCoV/YN2013, complete genome
Note 1c - Bat SARS-like coronavirus isolate bat-SL-CoVZC45, complete genome
Note2a - SARS coronavirus GZ0402, complete genome
Note 2b - SARS coronavirus isolate CFB/SZ/94/03, complete genome
Note2c - SARS coronavirus SZ3, complete genome
5. The
determining case of HIV1 Kenya 2008 absent from all coronaviruses other than
COVID-19 and bat RaTG13.
==> ==> Please see in Supplementary Materials (Ref 3) complete data on this particular EIE Kenya 2008. To summarize,
The case of HIV1 Kenya 2008
This important HIV1 genome was particularly studied in an HIV vaccine strategy context by Canadian Professor Franck Plummer Lab. Team [9], [10].
This region, in addition to its hundred strong homologies with all the COVID_19 strains of 2020, shows only two other homologies with, on the one hand, Bat coronavirus RaTG13, and at a lower level, with Rhinolophus affinis coronavirus isolate LYRa3 spike protein gene.
The HIV1 Kenya 2008 fingerprint recall: TGTTTTTATTACTTTTATTGCCACTATTCTCT
Here is the detail of these two main homologies:
Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome Sequence ID: NC_045512.2Length: 29903Number of Matches: 1
Score Expect Identities Gaps Strand
37.4 bits (40) 8e-04 28/32(88%) 1/32(3%) Plus/Plus
Query 1 TGTTTTTATTACTTTTATTGCCACTATTCTCT 32
||||||| || |||||||||||||| |||||
Sbjct 21568 TGTTTTTCTTG-TTTTATTGCCACTAGTCTCT 21598
Bat coronavirus RaTG13, complete genome
Sequence ID: MN996532.1Length: 29855Number of Matches1:
Score Expect Identities Gaps Strand
32.8 bits (35) 0.032 27/32(84%) 1/32(3%) Plus/Plus
Query 1 TGTTTTTATTACTTTTATTGCCACTATTCTCT 32
||||||| || |||||||||||||| | |||
Sbjct 21550 TGTTTTTCTTG-TTTTATTGCCACTAGTTTCT 21580
==> ==> Please, see the detailed Table 2.1 in Supplementary Materials Ref 4 (Dates of collection then deposit of various Bat genomes involved in the 225 bases area).
This Table results from the BLASTn analysis on April 10, 2020 option "SARS coronaviruses taxid 694009" reports 386 occurrences including 16 bats and 2 Rhinolophus, and 368 COVID_19.
In this Table, we demonstrate that in ALL Bats genomes others than Bat RaTG13 none of them have the presence of the EIE Kenya 2008.
In ALL cases, the 225 bases region is reduced to contiguous small regions between 17 and 96 bases length. In ALL cases, the Kenya 2008 EIE is totally absent.
We also note in this Table 6 that the Bats closest to COVID_19 were collected between 2013 and 2017, but only sequenced in 2020 (Bat RaTG13 (2013), Bat SARS-like coronavirus isolate Bat-SL-CoVZXC21 (2015), and Bat SARS-like coronavirus isolate bat-SL-CoVZC45 (2017). Alina Chan found that RaTG13 is the same as the “4991” strain with which Zheng-Li was working in 2017-18 (https://archive.vn/4Ot2j).
Location of the EIE HIV1 Kenya 2008 within the junction
between the 2 Orf1ab and Spike genes:
Firstly, the EIE regions of HIV1 Kenya 2008 nonfunctional (Sequence ID: EU875177.1) and of HIV1 Kenya real (Sequence ID: FJ623481.1) are identical while the respective Gp120 genes are only 82% homologous: 494/603 (82%).
HIV-1 isolate 06KECst_005 from Kenya, complete genome
Sequence ID: FJ623481.1Length: 8766Number of Matches: 1
Range 1: 5192 to 5794
|
Score |
Expect Identities |
Gaps |
Strand |
|
595 bits (659) |
6e-168 494/603(82%) |
3/603(0%) |
Plus/Plus |
The HIV1 Kenya EIE nonfunctional region from the COVID-19 genome is located overlapping between the end of the "Orf1ab" gene and the start of the "S spike" gene:
Details COVID-19 genes: Orf1ab Spike
266---------------21555 21563-----------------------------25384
HIV-1 Kenya 2008: 21542 21572
COVID_19 Wuhan market ID:LR757998.1 reference genome location of EIE Kenya 2008 HIV1: 21542-21572 bases.
Spike gene location: 21563-25384 bases.
So, in terms of amino acids:
START location of HIV1 KENYA: 21 amino acids before SPIKE begins.
END location of HIV1 KENYA: 9 amino acids after the beginning of SPIKE.
How about this same question in the case of bat RaTG13
genome?
The locations of HIV-1 Kenya within Bat RaTG13 Sequence ID: MN996532.1
is: 21550 TGTTTTTCTTG-TTTTATTGCCACTAGTTTCT 21580
(see RESULTS§ ref 3).
Location of the Spike gene within Bat RaTG13 is: 21545. 25354
/gene="S"
/codon_start=1
/product="spike glycoprotein"
/protein_id="QHR63300.2"
So, in terms of amino acids:
START address of HIV1 KENYA: 6 amino acids after SPIKE begins.
END address of HIV1 KENYA: 36 amino acids after the beginning of SPIKE.
Notably, unlike COVID-19 where HIV-1 Kenya starts before the start of the SPIKE gene, here, in the case of bat RaTG13, HIV1 Kenya is entirely contained within the SPIKE gene.
6. The
discovery of a new EIE from the HIV1 group «O» differentiating COVID-19 from
the Bat RaTG13 genome.
The HIV-1 group « O » constitutes a subgroup of HIV retroviruses very different comparing with others HIV/SIV subgroups, it appears particularly in Cameroon. However, little is known about group O and why this highly divergent retrovirus genome has not become pandemic [21].
We wanted to look for hypothetical traces of EIE coming from HIV group "O", more particularly, we looked for possible traces in COVID_19 and in bat RaTG13.
We then discover a POL (Integrase) homology from this strain HIV1 group "O", referenced as AF422215.1, which is located towards the 23800 bases of COVID_19.
==> On April 21, 2020, BLASTn reported 489 COVID_19 sequences - all the sequences available on this date - with ALL of the following homology: 20/22 (90.91%), except two2 high level deleted strains reported below.
==> As of May 4, 2020, BLASTn is providing 1578 COVID_19 sequences. All except 3 highly deleted at whole genome scale (Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/CA-CZB-IX00017/2020, ID: MT385497.1
, Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/UT-00087/2020,
ID: MT334549.1, Wuhan seafood market pneumonia virus genome, ID: LR757997.1) which are
very highly deleted contain this sequence completely preserved according to its homology of 20/22 bases, ie 90.91% of homology.
We must recall here this homology:
Between HIV-1 strain group O isolate 98CMA010 from Cameroon integrase (pol) gene, partial cds
GenBank: AF422215.1 https://www.ncbi.nlm.nih.gov/nuccore/AF422215.1
and
Wuhan seafood market pneumonia virus genome assembly, chromosome: whole_genome
Sequence ID: LR757998.1Length: 29866Number of Matches: 1
Range 1: 23804 to 23825
Score Expec Identities Gaps Strand t
31.9 bits (34) 3.0 20/22(91%) 0/22(0%) Plus/Plus
Query 532 ATGGCAGTATTTGTTCACAATT 553
|||||||| ||||| |||||||
Sbjct 23804 ATGGCAGTTTTTGTACACAATT 23825
The same research applied to Bat RaTG13 ID: MN996532.1 produces the results summarized by the Synthesis below:
|
Synthesis: |
|
|||
|
HIV1 Group O |
532 |
ATGGCAGTATTTGTTCACAATT 553 |
||
|
COVID_19 |
23804 |
ATGGCAGTTTTTGTACACAATT 23825 |
||
|
bat
RaTG13 |
23799 |
ATGGTAGTTTTTGCACACAATT 23820 |
||
|
differences |
|
X X between
COVID_19 and HIV1 gr O |
||
|
differences |
|
X X between COVID_19 and bat RaTG13 |
||
|
differences |
|
X X XX between
bat RaTG13 and HIV1 gr O (18/22) |
||
|
HIV1 Group O |
|
532 ATGGCAGTATTTGTTCACAATT 553 |
||
|
COVID_19 |
23804 |
ATGGCAGTTTTTGTACACAATT |
23825 |
|
|
bat
RaTG13 |
23799 |
ATGGTAGTTTTTGCACACAATT |
23820 |
|
|
bat-SL-CoVZXC21 |
23665 |
ATGGCAGTTTTTGCACACAA 23684 jui2015
/ |
5fev2020
/ 17/22 |
|
|
|
|
1 2 32 55 |
|
|
|
bat-SL-CoVZC45 |
23734 |
ATGGCAGTTTTTGCACACAA 23753 fev2017 / |
5fev2020
/ 18/22 |
|
|
|
|
1 2 32 55 |
|
|
|
SARS strain BtKY72 |
23639 |
ATGGTAGTTTCTGTACACAA 23658 aug2007 / |
8fev2020 / 17/22 |
|
|
|
|
3 4 12 55 |
|
|
|
Notes related to numbers
under sequences i.e 1,2,3,4,5: |
||||
Notes related to numbers under sequences i.e 1,2,3,4,5:
1) similar HIV1 group O see base T identical between HIV1 group « O » and SARS strain BtKY72 (note 1)
2) similar COVID_19 and bat RaTG13
3) similar bat RaTG13
4) different all (COVID_19 and bat RaTG13)
5) Absent contrarly HIV1 group O, COVID_19 and bat RaTG13
It is very interesting to note the following points:
· It is well known that bats have been studied in particular in China in recent years (https://en.wikipedia.org/wiki/Shi_Zhengli).
· The respective collection dates of these Bat genomes are 2007, 2013, 2015, 2017 while all of them were only sequenced in 2020 (with the exception of BtRf-BetaCoV / HeB2013, sequenced in 2017).
· We observe that all these Bat SARS strains have COVID_19 homologies in this region quite close to that of Bat RaTG13.
· It is remarkable to note (note1) this base T which is the only one to be simultaneously present in HIV1 group "O" and in SARS strain BtKY72.
· Finally, while COVID_19 has a homology of 20/22 bases with HIV1 group "O", Bat RaTG13 (2013) and bat-SL-CoVZC45 (2017) have a homology of 18/22 bases with HIV1 group "O".
7. Analysis
of local and global cohesions and heterogeneities of the 225 bases COVID_19,
bat RaTG13 and SARS Urbani genomes.
Now, we demonstrate how a new region including 4 HIV/SIV EIE radically distinguishes all COVID-19 strains from all SARS and Bat strains.
Then, we will be particularly interested in the Bat RaTG13 strain whose genomic proximity to COVID-19 will be analyzed with the greatest attention and precision.
The theoretical method used here makes it possible to evaluate the overall level of cohesion - then also of heterogeneity - of a sequence of nucleotides, and that independantly of the scale due to the fractal nature of this numerical method.
Full details on this numerical method in [6-8], and recall Methods in supplementary Materials ref 9.
Here we analyze the Master Code of 3 characteristic genomes COVID_19, bat RaTG13 and SARS Urbani.
We will study, for each of these 3 genomes, 5 successive amplitude scales and this according to the 3 reading frames of the codons and on the 2 main and complementary strands:
· whole genomes.
· bases 15,000 to 25,000.
· region including "A", "B", "Lyons Weiler".
· regions of 425 bases including 100, 225, 100 bases.
· 225 bases area.
Table 7: Synthetic Genomics/Proteomic global Master Code coupling (%). Note: we select in each case the best codons reading frame % coupling.
|
Genome |
Selective
Region 225 bases |
|
Wuhan
market ID: LR757998.1 |
69.47 |
|
Bat
RaTG13 ID: MN996532.1 |
92.13 |
|
SARS
Urbani ID: MK062180.1 |
Absent |
The main result to be discussed now is the comparison between both 225 bases area analyzes of COVID_19 and Bat RaTG13.
We must recall here both 225 bases area within Wuhan market ID: LR757998.1 reference and bat RaTG13 genomes:
Wuhan seafood market pneumonia virus genome assembly, chromosome: whole_genome
Sequence ID: LR757998.1Length: 29866Number of Matches: 1
Score Expect Identities Gaps Strand
407 bits(450) 7e-114 225/225(100%) 0/225(0%) Plus/Plus
Bat coronavirus RaTG13, complete genome
Sequence ID: MN996532.1Length: 29855Number of Matches: 1
Score Expect Identities Gaps Strand
312 bits (345) 4e-85 204/225(91%) 0/225(0%) Plus/Plus
The sequence SARS Urbani is totally absent selecting 1000 SARS like genomes in BLAST.
Homology of the 225 bases area between Wuhan market ID: LR757998.1 ref. and bat RaTG13 is very important: 204/225 bases (91% homology).
Analyzing the locations of the 4 HIV1 HIV2 EIE within the 225 bases area:
Wuhan market ID: LR757998.1 start address: 21543. Bat start address: 21550. Nucleotides and amino acids within Wuhan market ID: LR757998.1:
|
HIV1 Ken 471 501 |
ya
2008 Nucleotides
addresses within region « A » 600 bases |
|
1 31 |
Nucleotides
addresses within region 225 bases |
|
1 10 |
Amino
acids within region 225 bases |
|
HIV2
Cap verde 2012 512
529 Nucleotides addresses within region « A » 600 bases 42. 59 Nucleotides addresses within region
225 bases 14. 20 Amino
acids within region 225 bases |
|
HIV2 Cote d' ivoire 2014
66 85 Nucleotides addresses within region « B » 330 bases
195. 214. Nucleotides addresses within region 225 bases
65. 71 Amino acids within region 225 bases
SIV Africa 2016
76 97 Nucleotides addresses within region « B » 330 bases
205. 226 Nucleotides addresses within region 225 bases
68. 75 Amino acids within region 225 bases
Nucleotides homologies between Bat RaTG13 [21549 on 225 bases] and COVID_19 ID: LR757998.1 ref [21542 on 225 bases]
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 Kenya HIV1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 0 1 1 1 1 1 Cap verde HIV2
1 1 1 0 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 0 1 1 0 1 1 0 1 0
1 1 1 0 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1
1 1 1 0 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1
0 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1
0 1 1 2 last HIV2 and SIV have a
partial overlap.
1 1 1 1 1 1 1 1 1 1 1 0 1 1 1
Then, only 20 bases differences on 225 bases.
Note : The regions in bold correspond to the relative positions of the 4 EIEs HIV1 Kenya 2008, HIV2 Cape Verde 2012, HIV2 Cote d (ivoire 2014 and SIV Africa 2016. “1” significates same nucleotide value in COVID_19 and RaTG13. “0” significates different nucleotide value in COVID_19 and RaTG13.
Wuhan market ID: LR757998.1 ref region 225 basesFrame1
TGTTTTTCTTGTTTTATTGCCACTAGTCTC
TAGTCAGTGTGTTAATCTTACAACCAGAAC
TCAATTACCCCCTGCATACACTAATTCTTT
CACACGTGGTGTTTATTACCCTGACAAAGT
TTTCAGATCCTCAGTTTTACATTCAACTCA
GGACTTGTTCTTACCTTTCTTTTCCAATGT
TACTTGGTTCCATGCTATACATGTCTCTGG
GACCAATGGTACTAA
bat RaTG13 region 225 bases Frame1
TGTTTTTCTTGTTTTATTGCCACTAGTTTC
TAGTCAGTGTGTTAATCTAACAACTAGAAC
TCAGTTACCTCCTGCATACACCAACTCATC
CACCCGTGGTGTCTATTACCCTGACAAAGT
TTTCAGATCTTCAGTTTTACATTTAACTCA
GGATTTGTTTTTACCTTTCTTCTCCAATGT
GACCTGGTTCCATGCTATACATGTTTCAGG
GACCAATGGTATTAA
COVID_19 Wuhan market ID: LR757998.1 region 225 bases FRAME1
=======
CYS PHE SER CYS PHE ILE ALA THR SER LEU Kenya HIV1
ARR SER VAL CYS ARR SER TYR ASN GLN ASN Cap verde HIV2
SER ILE THR PRO CYS ILE HIS ARR PHE PHE
HIS THR TRP CYS LEU LEU PRO ARR GLN SER
PHE GLN ILE LEU SER PHE THR PHE ASN SER
GLY LEU VAL LEU THR PHE LEU PHE GLN CYS
TYR LEU VAL PRO CYS TYR THR CYS LEU TRP 2 last HIV1 and SIV have a partial overlap
ASP GLN TRP TYR ARR
bat RaTG13 region 225 bases FRAME1
=======
CYS PHE SER CYS PHE ILE ALA THR SER PHE Kenya HIV1
ARR SER VAL CYS ARR SER ASN ASN ARR ASN Cap verde HIV2
SER VAL THR SER CYS ILE HIS GLN LEU ILE
HIS PRO TRP CYS LEU LEU PRO ARR GLN SER
PHE GLN ILE PHE SER PHE THR PHE ASN SER
GLY PHE VAL PHE THR PHE LEU LEU GLN CYS
ASP LEU VAL PRO CYS TYR THR CYS PHE ARG 2 last HIV1 and SIV have a partial overlap
ASP GLN TRP TYR ARR
Note: The best nucleotides and amino acids matchings must be analyzed from the 3 codons and directions of codons reading frames.
In other words, in this above Table5 we see that apart from HIV1 KENYA the HIVs of the 225 bases area are more homologous in Wuhan market ID: LR757998.1 than in ba tRATG13.
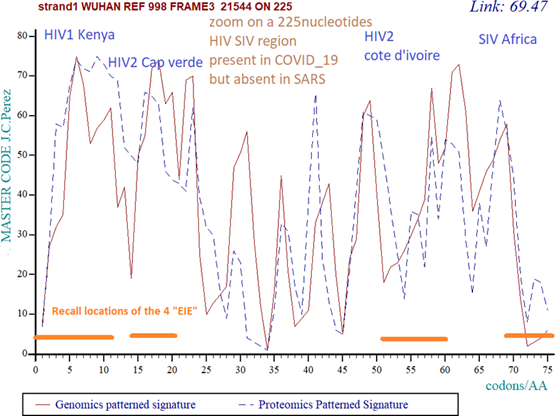
Figure 4: High level of HETEROGENEITY within the 225 bases area in Wuhan market reference genome. In this COVID_19 wuhan market ID: LR757998.1 reference genome, the coupling between Genomics pattern (red) and Proteomiics pattern (blue) appear highly disturbed, unstable, and “chaotic”. Their correlation is poor (69.47%).
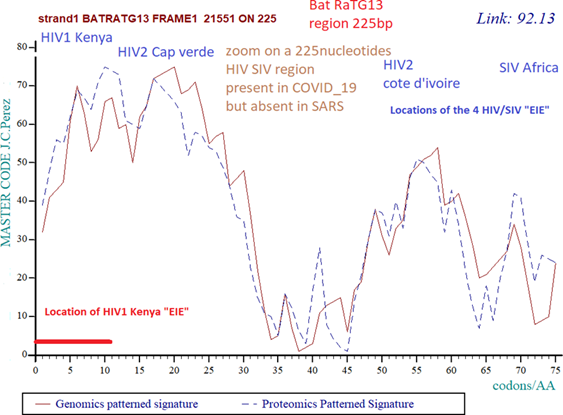
Figure 5: High level of COHESION in 225 bases bat RaTG13 region. This high level of COHESION in 225 bases bat RaTG13 region which include the fingerprint of Kenya HIV1 but, probably, not the 3 others HIV SIV signatures. Then, also, both Genomics pattern (red) and Proteomics pattern (blue) appear highly “harmonic” and correlated (92.13%).
We will draw the reader's attention to the 2 figs 4 and 5 above: The first concerns the 225 bases area of COVID-19 (Fig 4), it appears chaotic and not very organized. On the contrary, the same analysis for the same 225 bases region in bat RaTG13 (Fig 5) shows a more "smoothed" and regular profile. Let us not forget that this sequence, although filed in 2020, was taken in 2013, then 7 years earlier.
Here is how we explain this difference: the “DNA master code” (see supplementary materials ref 9) allows us to measure a certain level of cohesion and homogeneity between the genomic pattern (double stranded DNA) and its corresponding proteomic image (translation into amino acids). Here, as we pointed out in the article, the 3 EIEs cap verde, cote d'ivoire and Afrika were probably integrated by the natural evolution of Bat RaTG13, we would assume that the EIE Kenya would have has been integrated very recently (red line in Fig 5). On the contrary (Fig 4), for COVID_19, there are the whole 4 EIEs that would have been inserted very recently. This would result in this chaotic image in Fig 4.
Part III
In the decreasing slope of the epidemic, this 225 bases area on exhibits an abnormally high rate of mutations/deletions, particularly in USA Seattle WA state (§8, 9 and 10).
8. First
encouraging mutations in the 225 bases, « A » and « B » regions, particularly
in USA WA state.
We must recall here that the BLASTn analysis on April 10, 2020 option "SARS coronaviruses" reports 386 occurrences including 16 bats, 2 Rhinolophus, and 368 COVID_19. The same research running on 16 april 2020 reveals 523 strains sequences. The number of COVID_19 sequences available is therefore constantly changing principally due to USA new sequences deposits.
We were interested in the first cases of significant COVID_19 mutations in this key region of 225 bases (homologies of the order of 96%). we find 5 of them located in the BLASTn just in front of and near RaTG13, all come from the USA, taken and sequenced in April 2020, pathogenic.
A BLASTn analysis dated April 11, 2020 produces the following results: 386 sequences in total. whose:
351 strains with full 100% homology with 225 bases area.
17 strains with
mutations in 225 bases area.
18 strains bat.
Now let's look at these 17 cases of mutations in the 220 bases region.
Table 8: Mutations in region 225 bases
|
Strain
number |
Strain
reference |
Mutations
relatives addresses within 225 bases area |
Homologies |
HIV1/SIV EIE (note1) |
Collection
and deposit dates |
||||
|
1 |
USA |
SARS-CoV-2/WA-UW381/human/2020/USA, partial genome Sequence ID: MT263460.1 |
8 C/T |
224/225 99.6% |
HIV1 Kenya 2008 |
30
mar 2020 6
apr 2020 |
|||
|
2 |
USA |
SARS-CoV-2/WA- UW334/human/2020/USA, complete genome Sequence ID: MT263414.1 |
8 C/T |
224/225 99.6% |
HIV1 Kenya 2008 |
24 06 |
mar
2020 apr
2020 |
||
|
3 |
USA |
ARS-CoV-2/WA- UW301/human/2020/USA, complete genome Sequence ID: MT263384.1 |
81 C/T |
224/225 99.6% |
|
23 06 |
mar
2020 apr
2020 |
||
|
4 |
USA |
SARS-CoV-2/WA- UW270/human/2020/USA, partial genome Sequence ID: MT259262.1 |
79 C/T |
224/225 99.6% |
|
13 06 |
mar
2020 apr
2020 |
||
|
5 |
USA |
SARS-CoV-2/WA- UW257/human/2020/USA, complete genome Sequence ID: MT259249.1 |
157 |
G/C |
224/225 99.6% |
|
13
mar 2020 6
apr 2020 |
||
|
6 |
USA |
SARS-CoV-2/WA- UW231/human/2020/USA, complete genome Sequence ID: MT246488.1 |
8 |
C/T |
224/225 99.6% |
HIV1 kenya 2008 |
14 06 |
mar
2020 apr
2020 |
|
|
7 |
USA |
SARS-CoV-2/WA- UW204/human/2020/USA, complete genome Sequence ID: MT246461.1 |
8 |
C/T |
224/225 99.6% |
HIV1 kenya 2008 |
13 06 |
mar
2020 apr
2020 |
|
|
8 |
China |
SARS-CoV-2/KMS1/human/2020/CHN, complete genome Sequence ID: MT226610.1 |
217 |
T/A |
224/225 99.6% |
SIV
Africa 2016 |
20 06 |
jan 2020 apr 2020 |
|
|
9 |
Finland |
CoV-FIN-29-Jan-2020,
partial genome Sequence ID: MT020781.2 |
140 |
C/T |
224/225 99.6% |
|
29 17 |
jan
2020 mar
2020 |
|
|
10 China |
SARS-CoV-2/Yunnan- 01/human/2020/CHN, complete genome Sequence
ID: MT049951.1 |
77 T/A |
224/225 99.6% |
|
17 06 |
jan 2020 apr 2020 |
|||
|
11 USA |
2019-nCoV/USA-CA5/2020,
complete genome Sequence ID: MT027064.1 |
140 C/T |
224/225 99.6% |
|
24 mar 2020 06 apr 2020 |
||||
|
12 USA |
SARS-CoV-2/WA- UW302/human/2020/USA, partial genome Sequence ID: MT263385.1 |
175-176 CA/NN 164-166 CCT/NNN |
220/225 97.7% |
|
23 mar 2020 6 apr 2020 |
||||
|
13 USA |
SARS-CoV-2/WA- UW356/human/2020/USA, complete genome Sequence ID: MT263436.1 |
188-196 TTCCATGC T/NNNNNN NNN |
216/225 96% |
HIV2 cote d'ivoire 2014 |
24 mar 2020 06 apr 2020 |
||||
|
14 USA |
SARS-CoV-2/WA- UW351/human/2020/USA, complete genome Sequence ID: MT263431.1 |
189-197 TTCCATGCT A/NNNNNN NNN |
216/225 96% |
HIV2 cote d'ivoire 2014 |
24 mar 2020 06 apr 2020 |
||||
|
15 USA |
SARS-CoV-2/WA- UW287/human/2020/USA, complete genome Sequence ID: MT259277.1 |
189-197 TCCATGCT A/NNNNNN NNN |
216/225 96% |
HIV2 cote d'ivoire 2014 |
15 mar 2020 06 apr 2020 |
||||
|
16 USA |
SARS-CoV-2/WA- UW306/human/2020/USA, partial genome Sequence ID: MT263389.1 |
145-191 46 del |
144/144 100% then 34/34 |
|
23 mar 2020 06 apr 2020 |
||||
|
17
China |
Wuhan seafood market pneumonia virus genome assembly,
chromosome: whole_genome Sequence ID: LR757997.1 |
106-225 120 del |
1-105 100% |
HIV2 cote d'ivoire 2014 and SIV Africa 2016 |
31 dec 2019 06 mar 20209 |
||||
|
17 COVID-19 different strains ===> 5 different « IEE » HIV/SIV |
|||||||||
Note1: when the mutation is in HIV/SIV insert, we note the strain ref.
We observe that out of these 17 cases of mutations, the majority of them (13/17) concern the USA with dates posterior to the Chinese origin of the pandemic. Only 3 relate to China and one to Finland. There is probably the beginning of a mutation strategy of the genome to balance and integrate exogenous HIV EIE.
9 of these 17 mutations directly affect an HIV / SIV region. The others affect the intermediate region separating the 2 and 2 HIV / SIV pools.
It will also be noted that the majority of these strains come from recent samples (12/17 have dates of collection posterior or equal to March 2020). These dates would therefore correspond to a "mature" period of the COVID_19 genomes, which have now entered a phase of diversified mutations.
Finally, we observe the repetition of several mutations, proof of a robust mutation strategy which eliminates the hypothesis of sequencing errors.
We note that 5 different HIV/SIV EIE and 5 mutations regions are matching within the 17 different COVID_19 strains.
Now we consider Table 9 – Comparing 225 bases area significative mutations § deletions % with whole genomes mutations and deletions %.
Table 9: Comparing 225 bases area significative mutations § deletions % with whole genomes mutations and deletions %.
|
Strain number |
Strain reference |
Mutations relatives addresses |
Homologies
region 225 bass |
Homologies
whole genomes |
HIV1/S
IV EIE |
Collection and deposit |
|
|
|
within 225 bases area |
/ same
region in reference genome LR757998.1 and
mutations rate % |
/
whole reference
genome LR757998.1 and
mutations rate % |
|
dates |
|
12 USA |
SARS-CoV-2/WA- UW302/human/2020/USA, partial genome Sequence ID: MT263385.1 |
175-176 CA/NN 164-166 CCT/NNN |
220/225 97.7% 2.222222% |
29517/
29598 = 81 99.726333
% 0.273667% |
|
23 mar 2020 6 apr 2020 |
|
13 USA |
SARS-CoV-2/WA- UW356/human/2020/USA, complete genome Sequence ID: MT263436.1 |
188-196 TTCCATGC T/ NNNNNNN NN |
225-9 = 216 96% 4.000000% |
29828/
29846 = 18 99.939690
% 0.060309% |
HIV2 cote
d'ivoir e 2014 |
24 mar 2020 06 apr 2020 |
|
14 USA |
SARS-CoV-2/WA- UW351/human/2020/USA, complete genome Sequence ID: MT263431.1 |
189-197 TTCCATGC TA/NNNNN NNNN |
225-9 = 216 96% 4.000000% |
29834/
29852 = 18 99.939702
% 0.060297% |
HIV2 cote d'ivoir
e 2014 |
24 mar 2020 06 apr 2020 |
|
15 USA |
SARS-CoV-2/WA- UW287/human/2020/USA, complete genome Sequence ID: MT259277.1 |
189-197 TCCATGCT A/NNNNNN NNN |
225-9 = 216 96% 4.000000% |
29843/
29866 = 23 99.922989
% 0.077011% |
HIV2 cote
d'ivoir e 2014 |
15 mar 2020 06 apr 2020 |
|
16 USA |
SARS-CoV-2/WA- UW306/human/2020/USA, partial genome Sequence ID: MT263389.1 |
145-191 46 del |
225-179 = 46 79.5555% 20.44444% |
29517/
29598 = 81 99.726332
% 0.273667% |
|
23 mar 2020 06 apr 2020 |
|
17 China |
Wuhan seafood market pneumonia virus genome assembly,
chromosome: whole_genome Sequence ID: LR757997.1 |
106-225 120 del |
225-105 =120 46.6666% 53.333333% |
19263/29388 =
10125 65.547162
% 34.452838% |
HIV2 cote
d'ivoir e 2014 and |
31 dec 2019 06 mar 20209 |
|
|
|
|
|
|
SIV Africa 2016 |
|
In Table 9, results involving 6 significant genomes show a great average mutations level in each 225 bases regions (13.5687%) than in their relating whole genomes (0.3496%). Then a ratio between average rate mutations region 225 bases and average rate mutations whole genome = 38.813, due principally to the wuhan market hyper deleted genome LR757997.1
Note: last line ref17 China has many deleted or « N » regions: 19263 TCAG nucleotides on 29470 length, then 10207 nucleotides deletions or undetermined nucleotides regions.
The following Fig 6 illustrates these results.
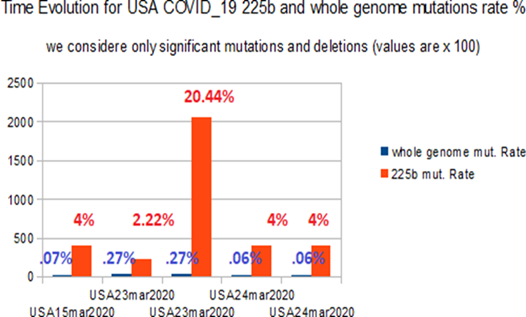
Figure 6: Comparative time evolution in WA mutations/deletions rates % at whole genome and 225 bases levels.
This chart illustrates for 5 COVID_19 USA strains collected from NCBI data banks in April 2020, the mutation rate from 225 bases regions and whole genomes. In all cases, the mutation rate is greater at 225 bases region that at whole genome scale.
Now, we do the same study for high density EIE regions « A » and « B » :
==> ==> The 2 Tables (Table Ref 6.1 and Table Ref 6.2) are available in Supplementary Materials Ref 6:
In Table Ref 6.1 – Region « A » interesting mutations, and in Table Ref 6.2 – Region « B » interesting mutations.
We obtain the same kind of results:
For region « A » analysis (Table Ref 6.1), we note that 5 different HIV/SIV EIE and 5 mutations regions are matching within the 8 different COVID_19 strains.
Supplementary Materials
For region « B » analysis (Table Ref 6.2), we note that 20 different HIV/SIV EIE and 13 mutations regions are matching within the 13 different COVID_19 strains.
Supplementary Materials
The following Fig 7 illustrates these highly significant results.
Fig 7 illustrates for 5 COVID_19 USA strains collected from NCBI data banks in April 2020, the mutation rate from regions « A »+ « B » (then 600+330bases) regions and whole genomes. In all cases, the mutation rate is greater at regions « A »+ « B » region that at whole genome scale.
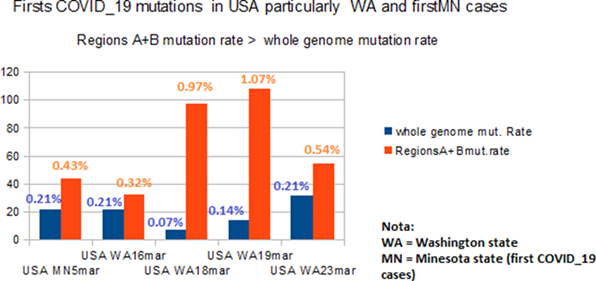
Figure 7: Comparative time evolution in WA / Minesota regions “A” and “B”. This chart represents (WA and Minesota strains first mutations) and mutations/deletions rates % at whole genome and in the case of region 930 bases = region « A » (600bases) + region « B » (330 bases).
Some conclusions on the geographical evolution of the genome:
In China, the strains seem to have changed very little in mutations (with the exception of Wuhan seafood market pneumonia virus genome assembly, chromosome: whole_genome Sequence ID: LR757997.1).
In Italy and in France, we find no remarkable mutation vis-à-vis the Chinese reference genome.
It is in Spain and the USA that we detect the most significant traces of a notorious evolution of the genome: In Spain, recent sequences (March 2020) demonstrate significant deletions and mutations in regions containing EIE. According to the first results of analyzes [13], this genome would not have increased its pathogenicity and would seem to use new modes of transmission.
In the USA, the analysis of multiple sequences from the Seatle region (WA) and Minnesota shows a clear growing trees progressiveness in the mutations then successive deletions of the regions "A", "B" and 225 bases, thus:
Table8 (ref 1 to 7, then 11 to 13), we progress from simple mutations to longer mutations on 3 codons, they affect HIV / SIV EIE.
Table Ref 6.1 (from Sup. Materials): also, there are grouped mutations (ref 4, 5) affecting EIE areas.
Table Ref 6.2 (from Sup. Materials): here we illustrate at best a sort of "shedding" of EIE regions in which these genomes progress: thus, (ref 3 5 6 7), the mutations affect 2 or 3, then 8 consecutive bases.
Then (9 10 11 12), in addition to other new mutations, it is whole pieces, on several tens of bases of the genome which are deleted. The most remarkable point is that in all these cases, it is indeed EIE regions which are targeted.
On the most recent date of April 23, 2020, we can check how other COVID_19 strains from Seatle WA have new deletions located in regions “A” and "B" of our article. It is deletions that are "shedding" in part of the EIE HIV / SIV located in region “A” and also in region “B”, particularly in the “side by side” EIE (see in Table 1: HIV1 Malawi 2013, HIV1 Russia 2010, SIV Cameroon 2015). There is the case particularly for:
Sequence ID: MT188341.1Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/WA-UW386/2020, partial genome
Length: 29835 collected 5mar2020, sequenced13mar2020,
Sequence ID: MT263466.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/WA-UW386/2020, partial genome
Length: 29634 collected 16mar2020, sequenced 15apr2020
Sequence ID: MT263385.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/WA-UW302/2020, partial genome
Length: 29610 collected 23mar2020, sequenced 15apr2020
Sequence ID: MT293224.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/WA-UW-1608/2020, complete genome
Length: 29847 collected 18mar2020, sequenced 15apr2020
Sequence ID: MT293213.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV- 2/human/USA/WA-UW-1574/2020, complete genome
Length: 29887 collected 19mar2020, sequenced 15apr2020
9. Generalization
of the analysis of 225 base regions in genomes of recent USA patients who have
mutated.
In order to formally demonstrate the specificity of this region of 225 bases located from base 21542 of 225 bases, we are exploring regions of the same size every 5000 bases throughout the genome of COVID_19. Let be from bases 1542, 6542, 11542, 16542, 26542. We can then deny or affirm the fact that this region of 225 bases that we have highlighted would indeed have a tendency to mutate or even to be partially deleted as this seems to appear for certain WA Seattle strains reported here (Fig 8). Table 10 below shows how the mutation rate of the 225 bases area is always much higher than that of the 5 regions 225 bases explored every 5000 bases (34.82 times).
Table 10: This Table summarizes remarkable results: they demonstrate the exclusive specificity of the 225 bases area which appears here in an obvious way to mutate in priority.
|
Strain numbe r |
Strain reference |
Mutation s relatives address es within 225 bases area |
Homologie s 225 bases area / same region in reference genome and mutations rate % |
Homologi es whole
genomes / whole reference genome and mutations rate % |
20kb Upstre am region 225 |
15kb Upstre am region 225 |
10kb Upstre am region 225 |
5kbUp strea m region 225 |
5kb Down strea m region 225 |
Ratio area 225 bases / avera ge 5 others 225 bases areas |
|
12 USA WA 23mar 2020 |
SARS-CoV- 2/WA- UW302/human /2020/USA, partial genome Sequence ID: MT263385.1 |
175-176 CA/NN 164-166 CCT/NN N |
220/225 97.7% 2.222222% |
29517/ 29598 = 81 99.72633 3 % 0.273667 % |
0,00% |
0,00% |
197 A/T 0.44% |
0,00% |
183- 185 CAC/N NN 1.33% |
6.24 Times |
|
13 USA WA 24mar 2020 |
SARS-CoV- 2/WA- UW356/human /2020/USA, complete genome Sequence |
188-196 TTCCAT GCT/ NNNNNN NNN |
225-9 = 216 96% 4.000000% |
29828/ 29846 = 18 99.93969 0 % 0.060309 % |
0,00% |
0,00% |
197 A/T 0.44% |
0,00% |
0,00% |
45 Times |
|
|
ID: MT263436.1 |
|
|
|
|
|
|
|
|
|
The following Fig 8 illustrates these strong results.
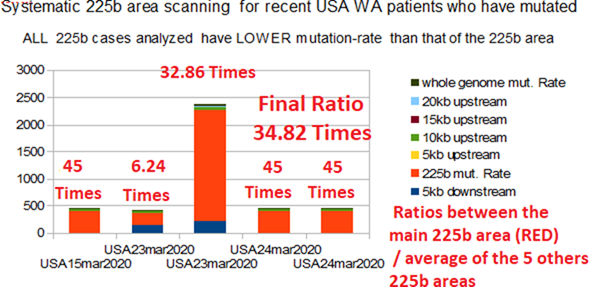
Figure 8: High level of deletions in the 225 bases area comparing to others 225 bases regions.
Horizontally: 5 patients from WA state with 225 bases area mutations. Vertically: proportional to mutations/deletions amount. The red surface is related to 225 bases Real area. The others four coloured areas are related to average amount of mutations/deletions rates for the 5 others 225 bases régions and whole genome. Ratio (i.e. 32.86 Times) is the ratio between the red 225 bases area and the average of others régions mutations/deletions rates. To summarize these remarkable results: they demonstrate (red areas) the exclusive specificity of the 225 bases area which appears here in an obvious way to mutate in priority, probably in order to get rid of the exogenous EIE regions characterizing this region.
10. New
evidence of increased deletions from region 225 bases in WA State in the USA.
As of May 2, 2020, we wanted to assess whether the 225 bases area of the COVID-19 strains continued to mutate in the WA state region in particular. Out of 1578 COVID_19 strains accessible to date, 32 presented significant mutations (more than 2 bases out of 225). Among them, 30 came from the USA (see table 12 below and Fig 9), the last 2 from Wuhan and the Czech Republic are not considered here. Among these 30 USA strains, 22 came from the state of WA, 5 from CA, 2 from Utah, and 1 from the state of New York.
The 3 most remarkable facts are:
On the one hand, a great diversity of places and types of mutations and deletions in the region of 225 bases. It will be interesting to locate these mutations vis-à-vis the positions of the 4 EIEs in this region.
On the other hand, new types of mutations are also appearing in states other than WA, in California in particular.We can conclude from this that this key region of 225 bases continues to be shed from its genome by the virus COVID_19.
Thirtly, there is a high variety and diversity of mutations and deletes: On these 30 USA cases, 20 cases are totally different mutation/deletions configurations.
Table 11: This Table demontrates expansion and diversity of 225 bases area on 2 May 2020, particularly in WA Seattle USA state.
|
Label |
Reference |
Strain
description |
Mutations/
deletions |
Mutations
rate |
Integrity
Genomic s/Proteo mics % Master Code |
|
|
USA0
WA |
Reference
Genome WA seattle |
Severe acute respiratory coronavirus 2 isolate2/human/USAWA-UW391/2020, genome |
Syndrome
SARS-CoV- complete |
0 del |
No |
88.4 |
|
|
|
GenBank:
MT293156.1 |
|
|
|
|
|
USA0
UT |
Reference
Genome UTah |
Severe acute respiratory coronavirus 2 isolate 2/Human/USA/UT-02025/2020, genome
Gerbante: MT536977.1 |
syndrome
SARS-CoV- complete |
0 del |
No |
84.7 |
|
USA0
NY |
Reference
Genome NY |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/NY-CDC-
SURV0985NYC/2020, complete genome Sequence ID: MT434817.1 |
0 del |
No |
86.5 |
|
|
CA1 |
USA
CA 28mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CA-CZB-
IX00112/2020, complete genome Sequence ID: MT385489.1 |
121
CAGAT/5N |
2.22% |
86.9 |
|
|
CA2 |
USA
CA |
Severe
acute respiratory syndrome coronavirus 2 |
164-166 |
2.22% |
51.8 |
|
|
|
28mar2020 |
isolate
SARS-CoV-2/human/USA/WA- |
CCT/NNN |
(1/5) |
|
|
|
|
|
UW302/2020,
partial genome Sequence
ID: MT263385.1 |
175-176 CA/NN |
|
|
|
|
WA1 |
USA
WA 23mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
2225/2020 ORF1ab polyprotein (ORF1ab) and ORF1a polyprotein (ORF1ab) genes,
partial cds; and surface glycoprotein (S), ORF3a protein (ORF3a), envelope
protein (E), membrane glycoprotein (M), ORF6 protein (ORF6), ORF7a protein
(ORF7a), ORF7b (ORF7b), ORF8 protein (ORF8), nucleocapsid phosphoprotein (N),
and ORF10 protein (ORF10) genes, complete cds Sequence ID: MT345837.1 |
177 ATGTTA/6N |
2.66% |
62.9 |
|
|
CA3 |
USA
CA 23mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CA-CZB-
EX00700/2020, complete genome Sequence ID: MT385494.1 |
137 TTACATTC/8N |
3.55% |
93.5
<== |
|
|
WA2 |
USA
WA 20mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1765/2020, complete genome Sequence ID: MT326134.1 |
189 TCCATGCTA/9
N |
4,00% |
85.9 |
|
|
WA3 |
USA
WA 20mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1698/2020, complete genome Sequence ID: MT326129.1 |
189 TCCATGCTA/9
N |
4,00% |
85.4 |
|
|
WA4 |
USA
WA 18mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1608/2020, complete genome Sequence ID: MT293224.1 |
188 TTCCATGCT/9
N |
4,00% |
87.1 |
|
|
WA5 |
USA
WA 19mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1574/2020, complete genome Sequence ID: MT293213.1 |
189 TCCATGCTA/9
N |
4,00% |
86 |
|
|
WA6 |
USA
WA 19mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1603/2020, complete genome Sequence ID: MT293200.1 |
189 TCCATGCTA/9
N |
4,00% |
86.8 |
|
|
WA7 |
USA
WA 19mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1583/2020, complete genome Sequence ID: MT293198.1 |
189 TCCATGCTA/9
N |
4,00% |
86 |
|
|
WA8 |
USA
WA 19mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1567/2020, complete genome Sequence ID: MT293196.1 |
189 TCCATGCTA/9
N |
4,00% |
85.8 |
|
|
WA9 |
USA WA 24mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-
UW356/2020, complete genome Sequence ID: MT263436.1 |
188 TTCCATGCT/9
N |
4,00%
(2/5) |
87.1 |
|
|
WA10 |
USA
WA 24mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-
UW351/2020, complete genome Sequence ID: MT263431.1 |
189 TCCATGCTA/9
N |
4,00%
(3/5) |
85.5 |
|
|
WA11 |
USA
WA 15mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-
UW287/2020, complete genome Sequence ID: MT259277.1 |
189 TCCATGCTA/9
N |
4,00%
(4/5) |
85.7 |
|
|
WA12 |
USA WA |
Severe
acute respiratory syndrome coronavirus 2 |
188 |
4,00% |
57.5 |
|
|
|
21mar2020 |
isolate
SARS-CoV-2/human/USA/WA-UW- 1758/2020 ORF1ab polyprotein (ORF1ab), ORF1a
polyprotein (ORF1ab), surface glycoprotein (S), ORF3a protein (ORF3a),
envelope protein (E), membrane glycoprotein (M), ORF6 protein (ORF6), ORF7a
protein (ORF7a), ORF7b (ORF7b), ORF8 protein (ORF8), nucleocapsid
phosphoprotein (N), and ORF10 protein (ORF10) genes, complete cds Sequence
ID: MT326171.1 |
TTCCATGCT/9
N |
|
|
|
|
WA13 |
USA
WA 24mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1963/2020 ORF1ab polyprotein (ORF1ab) and ORF1a polyprotein (ORF1ab) genes,
partial cds; surface glycoprotein (S), ORF3a protein (ORF3a), and envelope
protein (E) genes, complete cds; M gene, partial sequence; ORF6 gene,
complete sequence; and ORF7a protein (ORF7a), ORF7b (ORF7b), ORF8 protein (ORF8),
nucleocapsid phosphoprotein (N), and ORF10 protein (ORF10) genes, complete
cds Sequence
ID: MT326080.1 |
106-118 TTACCCTGAC
AAA/13N |
5.77% |
59.7 |
|
|
WA14 |
USA
WA 28mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
4749/2020, complete genome Sequence ID: MT375449.1 |
143-152 TCAACTCAG
G/10N 156
T/G 158
T/A 162
T/D 165
C/T |
6.22% |
83.1 |
|
|
CA4 |
USA
CA 8avr2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CA-CZB-
IX00141/2020, complete genome Sequence ID: MT385478.1 |
Del
32 bases 194-225 |
14.22% |
77.2 |
|
|
NY1 |
USA
NY 22mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/NY-
PV09161/2020 ORF1ab polyprotein (ORF1ab) gene, partial cds; ORF1a polyprotein
(ORF1ab) gene, complete cds; surface glycoprotein (S) gene, partial cds; and
ORF3a protein (ORF3a), envelope protein (E), membrane glycoprotein (M), ORF6
protein (ORF6), ORF7a protein (ORF7a), ORF7b (ORF7b), ORF8 protein (ORF8),
nucleocapsid phosphoprotein (N), and ORF10 protein (ORF10) genes, complete
cds Sequence
ID: MT371011.1 |
Del
32 bases 1- 32 |
14.22% |
63.1 |
|
|
WA15 |
USA
WA 27mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
4744/2020, complete genome Sequence ID: MT375448.1 |
166-178 TTTCTTTTCC
AAT/13N Del
12 214-225 |
11.11% |
71.7 |
|
|
CA5 |
USA
CA 25mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CA-CZB-
IX00017/2020, complete genome Sequence
ID: MT385497.1 |
125-144 AGATCCTCA
GTTTTACATT C/20N |
8.88% |
84.6 |
|
|
WA16 |
USA
WA 9mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate
SARS-CoV-2/human/USA/WA-UW71/2020, complete genome Sequence ID: MT252799.1 |
Del
42 bases 184-225 |
18.66% |
85.4 |
|
|
WA17 |
USA
WA 6avr2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
4707/2020, complete genome Sequence ID: MT375462.1 |
107-128 TACCCTGAC
AAAGTTTTC AGAT/22N |
9.77% |
67.8 |
|
|
WA18 |
USA
WA 16mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-
UW306/2020, partial genome Sequence |
Del
47 bases 145-191 |
20.88%
(5/5) |
67.2 |
|
|
|
|
ID: MT263389.1 |
|
|
|
|
|
WA19 |
USA
WA 20mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
1673/2020 ORF1ab polyprotein (ORF1ab) and ORF1a polyprotein (ORF1ab) genes,
partial cds; and surface glycoprotein (S), ORF3a protein (ORF3a), envelope
protein (E), membrane glycoprotein (M), ORF6 protein (ORF6), ORF7a protein
(ORF7a), ORF7b (ORF7b), ORF8 protein (ORF8), nucleocapsid phosphoprotein (N),
and ORF10 protein (ORF10) genes, complete cds Sequence ID: MT326131.1 |
Del
60 bases 132-191 220
A/N |
27.11% |
85.2 |
|
|
WA20 |
USA
WA 23mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
2220/2020 ORF1ab polyprotein (ORF1ab) and ORF1a polyprotein (ORF1ab) genes,
partial cds; surface glycoprotein (S) and ORF3a protein (ORF3a) genes,
complete cds; envelope protein (E) and membrane glycoprotein (M) genes,
partial cds; and ORF6 protein (ORF6), ORF7a protein (ORF7a), ORF7b (ORF7b),
ORF8 protein (ORF8), nucleocapsid
phosphoprotein (N), and ORF10 protein (ORF10) genes, complete cds Sequence
ID: MT345839.1 |
Del
53bases 129-181 |
23.55% |
69.8 |
|
|
UT1 |
USA
UT 25mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate
SARS-CoV-2/human/USA/UT-00302/2020 ORF1ab polyprotein (ORF1ab) gene, partial
cds; ORF1a polyprotein (ORF1ab) gene, complete cds; surface glycoprotein (S)
gene, partial cds; ORF3a protein (ORF3a) gene, complete cds; envelope protein
(E) and membrane glycoprotein (M) genes, partial cds; ORF6 protein (ORF6)
gene, complete cds; ORF7a protein (ORF7a) and ORF7b (ORF7b) genes, partial cds;
ORF8 protein (ORF8) gene, complete cds; nucleocapsid phosphoprotein (N)
gene, partial cds; and ORF10 gene, complete sequence Sequence
ID: MT334562.1 |
Del
99 bases 1- 99 |
44,00% |
74.4 |
|
|
CA6 |
USA
CA 31mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CA-CZB-
EX00719/2020, complete genome Sequence ID: MT385496.1 |
Del
102 bases 124-225 |
45.33% |
78.1 |
|
|
UT2 |
USA
UT 12mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/UT-00087/2020
ORF1ab polyprotein (ORF1ab), ORF1a polyprotein (ORF1ab), surface glycoprotein
(S), ORF3a protein (ORF3a), envelope protein (E), and membrane glycoprotein
(M) genes, partial cds; ORF6 protein (ORF6) gene, complete cds; ORF7a protein
(ORF7a) gene, partial cds; ORF7b gene, complete sequence; ORF8 protein (ORF8)
gene, partial cds; and nucleocapsid phosphoprotein (N) and ORF10 protein
(ORF10) genes, complete cds Sequence ID: MT334549.1 |
Del
103 bases 1-103 |
45.77% |
93.3
<== |
|
|
China1 |
China
Wuhan 31dec2019 |
Wuhan
seafood market pneumonia virus genome assembly, chromosome: whole_genome
Sequence ID: LR757997.1 |
Del
120 bases 106-225 |
53.33% (5) |
84.8 |
|
|
WA21 |
USA
WA 31mar2020 |
Severe
acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/WA-UW-
4582/2020, complete genome Sequence ID: MT375436.1 |
Del
190 bases 36-225 |
84.44% |
74.4 |
|
Note1 to Note5: these COVID_19 USA strains selected on our BLASTn April scanning (Table 9 and Fig 6) will be re-used, here, in Table11 and Fig 9. Then, we could compare 225 bases genome evolution and
increasing mutations rate between April and May BLASTn scanning analyzes, particularly in the cases of USA WA state COVID_19 strains.
Remark: Considering patients WA2 to WA12, we note 2 sets of common deletions (3 cases from base 188 collected 18 to 24 mars 2020, and 8 other cases from base 189 collected 15 to 24 Mars 2020). This Table 11 demontrates expansion and diversity of 225 bases area on 2 May 2020, particularly in WA Seattle USA state.
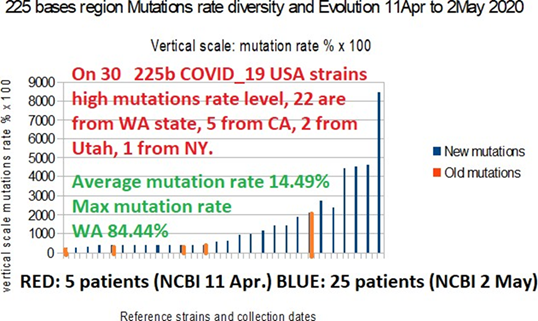
Figure 9: Analyzing mutations/deletions within 32 COVID_19 225 bases areas on 2 may 2020.
We compare evolution of patients with mutations/deletions between 2 NCBI genbank genomes sets collected with about 3 weeks delay. In "red" are the 5 "old" (11 April 2020) deletions from Table 10. In "blue" are the 25 "New" (2 May 2020) deletions from Table 11; we conclude that the COVID_19 genomes with deletions sequences available on 2 May 2020 has significantly increased in number but also in length of deletions. Then, we could conclude (blue colors) that USA COVID_19 genomes continue doing large deletions § mutations in critical 225 bases area. In the same time, both amount and diversity of these mutations are increasing and evolving.
Particularly, the average mutation rate of these 30 COVID_19 individual patients is 14.49% with a maximum WA state deletion case with 84.44% mutation rate.
Interestingly some of these deletions/mutations are touching the locations the 4 EIE present in this 225 bases area:
HIV1 Kenya 2008
1 31 Nucleotides addresses within region 225 bases
HIV2 Cap verde 2012
42. 59 Nucleotides addresses within region 225 bases
HIV2 Cote d' ivoire 2014
195. 214. Nucleotides addresses within region 225 bases
SIV Africa 2016
205. 226 Nucleotides addresses within region 225 bases
Locations of the 4 EIE within the 225 bases region (bold) within Wuhan market ID: LR757998.1 ref [21542 on 225 bases]
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 Kenya HIV1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 Cap verde HIV2
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 2 last HIV2 and SIV have a
partial overlap.
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
A detailed scanning of Table10 (Mutations/deletions column) reveals these intersting data:
Eleven (11) repeated cases of 9bases mutations are located between 188-197 or 189-198, then they « cut » the final HIV/SIV region starting in base 195. Others big deletions destroys systematically the 2 starting EIE region (1-59) or the 2 end EIE region (195-225): i.e Del 32 bases 194-225 and Del 32 bases 1-32 (which destroys exactly HIV1 Kenya EIE). Others bigger deletions erase half (begin or end) sections of the 225 bases region: i.e Del 99 bases 1-99, Del 102 bases 124-225 etc...
Finally, in 20 cases on 30 analyzed USA patients
mutations/deletions affect partially or totally one or more of the 4 HIV/SIV
EIE regions.
Part IV
The comparative analysis of the SPIKES genes of COVID_19
and Bat RaTG13 (§11, 12, 13 and 14).
11. The
region 1770 bases of the 2 proteins SPIKE in COVID_19 and Bat RaTG13.
We will be interested in the sequences of the 2 respective SPIKE proteins of COVID_19 (reference genome used in the article) and Bat RaTG13.The relative addresses are respectively: SPIKBAT: address in Bat RaTG13 of address 21545 on 3810 bases.
SPIKCOV: address in COVID_19 (ref 998) of address 21538 on 3822 bases.
The comparative analysis of these 2 SPIKES sequences highlights the following partition: 1-A first region between bases 1 and 2040 common to COVID_19 and bat RaTG13.
Then, for Spike COVID_19 only, an insertion of 12 bases (CCTCGGCGGGCA) corresponding to the 4 amino acids "PRRA" (Pro, Arg, Arg, and Ala).
Then comes a second common region of 1,770 bases: Located from 2041 on 1770 bases for Bat RaTG13. And located from 2053 to 1770 bases for COVID_19.
We are then confronted with two “anomalies" which are dificult to explain in natural biological conditions:
1) A short insert of 4 amino acids PRRA. This insert is UNIQUE in COVID_19 and does not exist in Bat RaTG13.
2) When comparing for these 2 pairs of regions the synonymous mutations and the non synonymous mutations, an abnormal fact will be highlighted for the second of the regions, that
The first region of 2040 bases (680 amino acids) common to the SPIKES of COVID_19 and Bat RaTG13:The 2 sequences are differentiated by 172 nucleotide mutations.
Let’s finally:
155 different codons.
101 synonymous codons.
For 54 non-synonymous codons.
Then a ratio “Codons synonyms” / “Codons not synonyms” = 101/54 = 1.8703.
Therefore,
“bases involved in synonymous codons” / “bases involved in not synonymous codons” = 5.611. This value close to the ratio “5” corresponds to the standard usually encountered in natural genetic sequences.
The second region of 1770 bases (590 amino acids) common to the SPIKES of COVID_19 and Bat RaTG13: The 2 sequences are differentiated by 90 nucleotide mutations.
Let’s finally:
89 different codons.
83 synonymous codons.
For 6 non synonymous codons ONLY.
Either a ratio “Codons synonyms” / “Codons not synonyms” = 83/6 = 13.8333
Therefore,
“bases involved in synonymous codons” / bases involved in not synonymous codons”= 41.499 .
Thus down tream PRRA region (41.499) is 7.396 times
greater than upstream PRRA region (5.611).
This 1770b region represents an "abnormal" level because the ratio of synonymous codons / non- synonymous codons = 41 is completely ABNORMAL. This suggests the possible manipulation of this region of the COVID_19 genome.
Fig 10 below illustrates these “abnormal” results.
And it is the following § which will bring us an unexpected
answer to this question ...
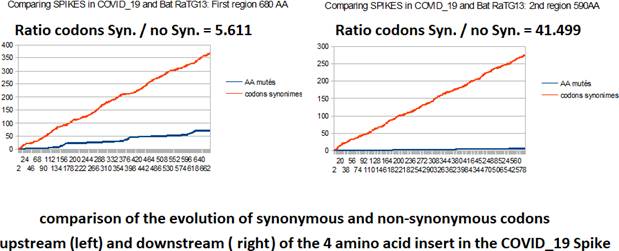
Figure 10: Comparing all codons mutations differenciating both Spikes related to COVID_19 and Bat RaTG13.
On the left, we represent the 2040b Spike region upstream the 4 amino acids insert, on the right we represent the 1770b region downstream the 4 amino acids insert. In red, the synonymous codons, in blue the non-synonymous codons. The right chart appears “unnatural”.
It is agreed that covid_19 would come from bat RaTG13. In such a case, the codons of covid_19 would have been modified from those of bat RaTG13.
The majority of these mutations would have led to synonymous codons whereas only 6 out of 590 amino acids in the 1770 base region would have changed values, or around 1%, which remains very low. A question then remains open: why this very low number of mutations in non synonymous codons?
Let us try to explain this abnormal phenomenon. When
mutations are natural, the rate of synonymous/ non- synonymous codon mutations
is close to 5. This is the case for the region of 2040 bases located upstream
of the PRRA (left image in Fig 10.). What is abnormal in the right part of Fig
10., region 1770b, is the very low number of non-synonymous codons (blue)
because the rate of change of synonymous codons is normal: the slopes of the 2
straight lines in red are similar. But, paradoxically, it is in the variation
of synonymous codons that an explanation of the anomaly must be sought. In Fig
11. of next & 12, we demonstrate that almost all of the nucleotide mutations
of this region 1770b concern the third base of codons, precisely, that which
generally does not change the amino acid and produces a synonymous codon. The
only question we will not be able to answer will be this, a question of ANTERIORITY:
"were the 1770 bases region abnormal mutations of
synonymous codons carried out on COVID_19 or on RaTG13?"
An exhaustive inventory of synonymous mutations: « how
did 89 codon mutations only lead to six amino acid changes? »
We sought, in particular, the distribution of mutations on the 3rd bases of the 84 synonymous codons: 77 of these 84 codons are divided into 3 classes:
1) class 1. 42 TC or CT.
2) class 2. 18 AG or GA.
3) class 3. 17 TA or AT.
Classes 1 and 2, i.e. 60 mutations, are of the transitions type:(Transition:each of the 4 nucleotide changes between purines or between pyrimidines: T <=> C or A <=> G).
Whoever observes the structure of the table of the universal genetic code organized according to the TCAG order, will notice that the 60 codons of classes 1 and 2 are found in 2 adjoining vertical boxes, therefore in the same amino acid. Likewise, certain amino acids like GLY, VAL, PRO, LEU, SER, ALA, THR or ARG occupy 4 contiguous vertical cells, where the 17 mutations of class3 TA/AT produce the same amino acid.
This is how we demonstrate how 77 of 84 mutations on the 3rd base of codons will not have produced amino acid changes.
12. Evidence
of a SPIKE significant EIE of Plasmodium Yoelii and of a possible HIV1 EIE with
a crucial Spike mutation.
The search for possible EIEs in COVID_19 and Bat RaTG13, both at the level of whole genomes, of the protein Spike, or of the critical region of 1770 bases highlights different candidate EIEs (see supplementary materials ref 7). The analysis of the region of 1770 bases more particularly reveals an EIE with a high probability BLASTn, moreover, the analysis via the Master Code points to a very probably precise functional site in this same region located towards the relative address 300 (100 amino acids (see supplementary materials ref 7a):
Plasmodium yoelii strain 17X genome assembly, chromosome: 10
Sequence ID: LM993664.2Length: 2065729Number of Matches: 2
Score Expect Identities Gaps Strand
46.4 bits (50) 0.004 36/42(86%) 1/42(2%) Plus/Plus
Query 296 CACAAGTCAAACAAATTTACAAAACAC-CACCAATTAAAGAT 336
||||| ||||||||||||||||||||| ||||| ||| || Sbjct 5556
CACAAATCAAACAAATTTACAAAACACAAACCAAAAAAAAAT 5597
This EIE appears in several chromosomes of the plasmodium yoelii. In particular, it was quickly identified as a protein with the name “Fam a” Plasmodium yoelii “fam-a” protein (PY17X_0018000), partial mRNA Sequence ID: XM_022956016.1
We should remember here that Plasmodium Yoelii is studied in mice in malaria vaccine
strategies [29].
An analysis of amino acid homologies confirms the very probable insertion of this EIE in COVID_19, in fact, 10 amino acids concentrated in a short sequence are homologous between COVID_19 and Plasmodium Yoelii protein "Fam a" (supplementary materials ref7b).
Analysis of the region in SPIKE Covid_19, located at the address 2052 + 295 on 42 product bases:
CAC AAG TCA AAC AAA TTT ACA AAA CAC CAC CAA TTA AAG ATT …/...
Either on the first reading frame of the codons:
HIS LYS SER ASN LYS PHE THR LYS HIS HIS GLN LEU LYS ILE …/...
We can easily verify that this codon reading frame is indeed that of the "Fam a" protein:
/product="fam-a protein protein_id="XP_022810934.1"
/db_xref="GeneID:3801450"
/translation="MNIFFVQIVLFLLIISLCVNKNTLATELIPKKDKK
HKSNKFTKH KP
K
KNKKCYPTYDNTKEIYQKN.../...
The homologous region on yoelii "Fam a", produces:
CAC AAA TCA AAC AAA TTT ACA AAA CAC AAA CCA AAA AAA AAT.../...
Either on the first reading frame of the codons:
HIS LYS SER ASN LYS PHE THR LYS HIS LYS PRO LYS LYS ASN.../...
Or an almost perfect homology of amino acids despite 2 synonymous codons underlined here (AAG / AAA and AAG / AAA).
For information, the same analysis conducted on Bat RaTG13 produces:
CTC AAG TTA AAC AAA TTT ATA AGA CAC CAC CAA TTA AAG ATT …/...
LEU LYS LEU ASN LYS PHE ILE ARG HIS HIS GLN LEU LYS ILE …/...
The remarkable fact is the following: the amino acid homology between the region COVID_19 and Yoelii "Fam a" (10/14) is greater than that between Bat RaTG13 and yoelli "Fam a" (6/14), and equivalent to the homology between Bat RaTG13 and COVID_19 (10/14).
Which is much less obvious as homology (6 amino acids instead of 10).
One question: did this Plasmodium yoelii EIE already exist in SARS? We analyze SARS Exon1 Sequence ID: FJ882956.1 (collected 2008, sequenced then published 2010). Curiously, another small homology with SIV ENV appear also (see supplementary materials ref 7c and ref7d).
The following cross homologies with Plasmodium Yoelii quickly appear:
SIV 24/33 bases 3/14 amino acids.
SARS. 31/42 bases. 8/14 amino acids (including a Stop codon). Bat RaTG13. 34/42 bases 6/14 amino acids.
COVID_19. 36/42 bases 10/14 amino acids.
Finally, the global homology between these 5 sequences is:
SARS CTC AAG TCA AAC AAA TGT ACA AAA CCC CAA CTT TGA AAT ATT RATG13 CTC AAG TTA AAC AAA TTT ATA AGA CAC CAC CAA TTA AAG ATT COVID CAC AAG TCA AAC AAA TTT ACA AAA CAC CAC CAA TTA AAG ATT YOELII CAC AAA TCA AAC AAA TTT ACA AAA CAC AAA CCA AAA AAA AAT
SIV AC AAG gCA AA_ AgA gTT AgA AAA CAC CAC CAA T...
Meanwhile, the homology between COVID_19 and SIV is here:
SIV / COVID_19: 28/33 bases 5/14 amino acids.
In this array we underlined amino acids homologies. It can be seen in this table that the amino acids of COVID-19 homologous to those of Yoelii result from a sort of "fusion" between those of SARS and those of Bat RaTG13.
It is interesting to note that this EIE of Plasmodium Yoelii in Spike COVID_19 is not an isolated case. For example, in the region "B" of 330 bases, very rich in EIE HIV / SIV, we can demonstrate the presence of EIE of Plasmodium Yoelii proteins (see supplementary materials ref 7e).
Another homology is added: SIV (supplementary materials ref 7d):
Simian immunodeficiency virus isolate UG31 from Tanzania gag protein (gag) and pol polyprotein (pol) genes, partial cds; vif protein (vif) and vpr protein (vpr) genes, complete cds; and tat protein (tat), rev protein (rev), and envelope glycoprotein (env) genes, partial cds
Sequence ID: JN091692.1Length: 5254Number of Matches: 1
Score Expect Identities Gaps Strand
34.6 bits(37) 7.8 28/33(85%) 1/33(3%) Plus/Plus
Query 297 ACAAGTCAAACAAATTTACAAAACACCACCAAT 329
||||| |||| | | ||| ||||||||||||||
Sbjct 2232 ACAAGGCAAA-AGAGTTAGAAAACACCACCAAT 2263
Another question: does this homology between COVID_19 and "Fam a" continue beyond? Indeed, an apparent continuity of this protein located downstream would extend this homology over a length of more than 60 bases:
Plasmodium yoelii genome assembly PYYM01, chromosome : 14
Sequence ID: LK934642.1Length: 2614191Number of Matches: 1
Score Expec Identities Gaps Strand t
41.9 bits(45) 0.16 42/54(78%) 2/54(3%) Plus/Minus
uery 309 AATTTA--CAAAACACCACCAATTAAAGATTTTGGTGGTTTTAATTTTTCACAA 360
|||||| ||||| | |||||||| | |||||| | | ||||||||||| ||
Sbjct 1561202 AATTTAGTCAAAATAAAACCAATTATATATTTTGATCATATTAATTTTTCAAAA 1561149
In [27], we had already demonstrated the presence of several EIEs of plasmodium yoelii in the "Lyons weiler" region of COVID_19. Indeed, thanks to a method allowing to detect heterogeneous sequences, therefore can be exogenous, we had suspected the possible presence of such sequences in the region "Lyons weiler” (& 7 and Figs 2 and 3 in [27]). By re-visiting this region, we show the existence of at least 4 EIEs in this region of COVID_19 Spike "Lyons weiler" région addresses 219, 464, 689, e 1132 (see supplementary materials ref 7f). In June 2020, a Korean team has just confirmed our results by publishing a PREPRINT demonstrating the presence of homologous sequences to Plasmodium in this same region [28].
Finally, here is the alignment of the nucleotides of these 3 respective sequences: COVID_19, Bat RaTG13 and Yoelii "Fam a":
COVID19 CACAAGTCAAACAAATTTACAAAACACCACCAATTAAAGATTTTGGTGGTTTTAATTTTTCAC RATG13 CTCAAGTTAAACAAATTTATAAGACACCACCAATTAAAGATTTTGGTGGTTTCAATTTTTCAC
YOELII CACAAATCAAAAATTTAGTC AAAATAAAACCAATTATATATTTTGATCATATTAATTTTTCAA
Note: The underlined part in yoeli comes from the second yoelii fragment of this second Blastn.
COVID_19 on 63 bases:
CACAAGTCAAACAAATTTACAAAACACCACCAATTAAAGATTTTGGTGGTTTTAATTTTTCAC
HIS LYS SER ASN LYS PHE THR LYS HIS HIS GLN LEU LYS ILE LEU VAL VAL LEU ILE PHE HIS
RaTG13 on 63 bases: CTCAAGTTAAACAAATTTATAAGACACCACCAATTAAAGATTTTGGTGGTTTCAATTTTTCAC
LEU LYS LEU ASN LYS PHE ILE ARG HIS HIS GLN LEU LYS ILE LEU VAL VAL SER ILE PHE HIS
Yoelii « Fam a » on 63 bases :
CACAAATCAAAAATTTAGTCAAAATAAAACCAATTATATATTTTGATCATATTAATTTTTCAA
HIS LYS SER LYS ILE ARR SER LYS ARR ASN GLN LEU TYR ILE LEU ILE ILE LEU ILE PHE GLN
Therefore, the relative homologies in nucleotides, then in amino acids over this length extended to 63 bases, that is to say 21 amino acids lead to:
COVID_19 / Bat RaTG13 = 58/63b et 16/21AA
COVID_19 / Yoelii « Fam a » = 46/63b et 11/21AA Bat RaTG13 / Yoelli « Fam a » = 41/63b et 7/21AA
It is therefore clear that this second region of Yoelii does not coincide with the extension downstream of the sequence "Fam a", although concatenated with the fragment Yoelii "Fam a" in COVID_19, this region would come from another region (functional ?) from Plasmodium Yoelii ...
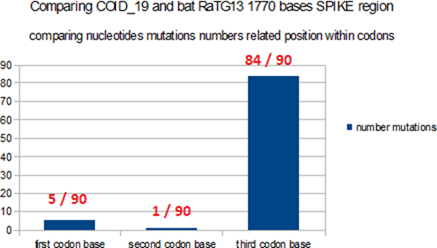
Figure 11: Comparing bases codons positions in COVID_19 and Bat RaTG13 1770 bases SPIKE region.
Evidence that the majority of the 90 nucleotide mutations between COVID_19 and Bat RaTG13 SPIKE region 1770 bases are located on the third bases of the codons.
It will be interesting to note this major fact: in [26] (Fig 1), Petrovski et al demonstrate a whole region where the amino acids are massively changed between SARS and COVID_19. Very precisely, this region is the region of 1770 bases of u SPIKE of COVID_19 where the amino acids are almost ALL IDENTICAL between COVID_19 and Bat RaTG13, whereas, at the same time, almost all the codons are c "changed" into synonymous codons.
The major conclusion of this demonstration of an EIE of the plasmodium Yoelii in COVID_19 is as follows: This very high amino acid homology score of 10/14 between covid / yoelii "Fam a" results from a shift in the reading frame of the spike codons. This explains the poorer score of the RaTG13 bat with respect to the yoelii which, however, is homologous in amino acids in this region which is very poor in amino acid mutations! So these are the basic mini mutations between COVID_19 and bat RaTG13 which made the difference here!
With this proof of yoelii, we obtain at the same time the explanation of this anomaly of the ratio codons synonyms / non-synonyms of the region 1770b highlighted previously. Indeed, as shown in Fig 11 above, the minor mutations do not change the amino acid values COVID_19 / bat RaTG13 (almost always the 3rd base of synonymous codons).
We believe that this strategy of shifting the codon reading frame was probably used throughout this region of 1770 bases, for example in this location (relative to 1770 bases region):
1464 TAATGCTTCAGTTGTAAA-CATTCAAAAA 1491 with 93% nucleotides homology, and a good amino acids homology considering the shift of codons reading frame. Effectively, this other EIE from plasmodium Yoelii also corresponds to a shifted position from the reading frame for Spike codons (see supplementary materials).
But with the change of the codon reading frame, a “synonymous” mutation on the Spike frame will become “not synonymous” on a second codon reading frame, which has just been demonstrated here, this is very precisely what who arrives here with this blatant proof of the fact that an EIE of the gene "Fam a" of the plasmodium Yoelii would have been inserted here using this "strategy for intelligent": while the 2 genes SPIKE of COVID-19 and Bat RaTG13 are almost identical according to their normal reading frame, a second reading frame radically differentiates the expression of the EIE "Fam a" between the 2 respective Spikes of COVID_19 and Bat RaTG13.
A possible HIV1 EIE contains a crucial Spike mutation.
Besides this EIE of plasmodium yoelii, it seems important to note this other smaller and hypothetical EIE in the region 2040b (S1) of the Spike.
We analyze the region 1801 to 1899 of Spike, its 33 amino acids contain an important mutation of Spike.
GGAACAAATACTTCTAACCAGGTTGCTGTTCTTTATCAGGATGTTAACTGCACAGAAGTCCCTGTTGCTATTCATGCAGA TCAACTTACTCCTACTTGG
End of April 2020, Bette Korber, from the Los Alamos National Laboratory, in New Mexico, claimed that a strain carrying a mutation called S-D614G seemed to take precedence over the others when it competed in a given geographic territory.
In vitro studies at the Scripps Research Department of Immunology and Microbiology of Florida have just confirmed this theory today. When they had this mutation, viruses more easily infected human cells in vitro [32].
This mutation identified in early March in Europe, Mexico, Brazil and China, Wuhan, modifies the structure of the Spike protein. This mutation, S-D614G: a glycine GLY replaced an aspartic acid ASP on codon 614 of protein Spike.
HIV-1 M:08GQ267 partial pol gene for gag-pol fusion polyprotein precursor, isolate 08GQ267
Sequence ID: FN557340.1Length: 1751Number of Matches: 1

If we make the mutation GAT (ASP) ==> GGT (GLY)
This EIE homology with HIV1 is lost.
COVID_19 becomes active if protein S is separated by an enzyme in S1 and S2 which then become functional, without however completely detaching from each other. It's here that the mutation acts: it seems to make the bond more "stable"
linking S1 and S2 after action of this enzyme.
The mutation "stabilizes" the virus in its most form effective.
This would explain the predominance of this mutated strain. The mutation is present in 70% of the samples posted on Genbank in May 2020, and it now epresents 60% of the strains present in Genbank. This strain has circulated a lot in France, Italy and now in the USA, but almost not in the State of WA studied in our article. If we do not find deletions of this strain in WA, Genbank contains strains where this area is deleted in other places: Australia, India, USA MAsachussets, CAlifornia, UTah, and especially FLorida.
As we have shown for other areas of the genome (WA state Seattle), it seems that, here too, the genome is trying to delete this region of the Spike.
13. The
analysis of deletions in the SPIKE critical region of 1770 bases in the USA WA
state (Seattle).
As we did above for the region 225 bases of COVID_19, we will ask ourselves here the same question: "The region of 1770 bases of Spike, and more particularly the EIE of Plasmodium Yoelii undergo strong deletions in genomes from USA patients from Washington State WA Seattle "?
Table 12: 23 USA” WA state” individual patient genomes with deletions in the 1770 bases COVID_19 SPIKE region.
|
|
23
USA WA individual patient partially deleted whole genome |
Deletions |
Plasmodi
um Yoelii deletions |
Genomics/
Proteomics % COUPLING |
|
Reference
Genome WUHAN998 |
Wuhan seafood market
pneumonia virus genome assembly, chromosome: whole_genome ref LR757998.1. |
0 del |
No |
86.2 |
|
Reference
Genome WA seattle |
Severe acute respiratory
syndrome coronavirus 2 isolate SARS-CoV- 2/human/USAWA-UW391/2020, complete
genome GenBank:
MT293156.1 |
0 del |
No |
88.4 |
|
WA77 |
USA/WA-UW-5205/2020,
complete genome Sequence ID: MT412257.1 |
6 del |
No |
87.3 |
|
WA78 |
USA/WA-UW-5182/2020,
complete genome Sequence ID: MT412228.1 |
6 del |
No |
84.6 |
|
WA79 |
USA/WA-UW146/2020,
complete genome Sequence ID: MT252737.1 |
8 del |
No |
80.7 |
|
WA80 |
USA/WA-UW273/2020,
partial genome Sequence ID: MT259265.1 |
8 del |
No |
68.6 |
|
WA81 |
USA/WA-UW199/2020,
complete genome Sequence ID: MT246456.1 |
13del |
No |
65.4 |
|
WA82 |
USA/WA-UW280/2020,
partial genome Sequence ID: MT259272.1 |
18del |
No |
61.2 |
|
WA83 |
USA/WA-UW302/2020,
partial genome Sequence ID: MT263385.1 |
21del |
No |
51.8 |
|
WA84 |
USA/WA-UW373/2020,
complete genome Sequence ID: MT263453.1 |
25del |
296-300 |
75. |
|
WA85 |
USA/WA-UW386/2020,
partial genome Sequence ID: MT263466.1 |
33del |
Close
upstream |
74.3 |
|
|
|
|
Yoelii |
|
|
WA86 |
USA/WA-UW278/2020,
partial genome Sequence ID: MT259270.1 |
38del |
No |
66.2 |
|
WA87 |
USA/WA-UW306/2020,
partial genome Sequence ID: MT263389.1 |
39del |
No |
67.2 |
|
WA88 |
USA/WA-UW206/2020,
partial genome Sequence ID: MT246463.1 |
44del |
301-313 and
322- 336 |
82. |
|
WA89 |
USA/WA-UW289/2020,
partial genome Sequence ID: MT259279.1 |
45del |
301-313 and
close downstre am Yoelii |
74.4 |
|
WA90 |
USA/WA-UW-6315/2020,
complete genome Sequence ID: MT412323.1 |
46del |
301-313 and 332-336 |
74/4 |
|
WA91 |
USA/WA-UW208/2020,
partial genome Sequence
ID: MT246465.1 |
66del |
301-313 and 320-326 and
330- 336 |
69.6 |
|
WA92 |
USA/WA-UW312/2020,
partial genome Sequence
ID: MT263393.1 |
99del |
No |
75.7 |
|
WA93 |
USA/WA-UW-4538/2020,
complete genome Sequence
ID: MT375428.1 |
129del |
Totally
deleted |
81.6 |
|
WA94 |
USA/WA-UW347/2020,
partial genome Sequence
ID: MT263427.1 |
198del |
Totally
deleted |
57.1 |
|
WA95 |
USA/WA-UW157/2020,
complete genome Sequence
ID: MT252730.1 |
167del |
Totally
deleted |
85.8 |
|
WA96 |
USA/WA-UW-4707/2020,
complete genome Sequence
ID: MT375462.1 |
180del |
No |
67.8 |
|
wa97 |
USA/WA-UW379/2020,
partial genome Sequence
ID: MT263409.1 |
361del |
Totally
deleted |
74. |
|
WA98 |
USA/WA-UW246/2020,
partial genome Sequence
ID: MT259238.1 |
413del |
322-336 |
78.2 |
|
WA99 |
USA/WA-UW267/2020,
partial genome Sequence
ID: MT259259.1 |
390del |
Totally
deleted |
84.2 |
|
|
Summary |
23 deletions / 23
cases |
12 undelete
d, 6 partially
deleted, 5
totally deleted |
23 on
23 have a lower % than WA state
COVID_19 reference |
Note: we have selected here the last 23 WA (Seattle) genomes resulting from a BLASTn search carried out on the 1770 bases region on the GENBANK COVID_19 sequences public database on May 27, 2020.
Complete details in supplementary materials (ref 8).
It appears here very clearly that these genomes of the USA WA state (Seattle) region seem to try to "rid" of these EIE regions: indeed, of these 23 genomes analyzed, almost half have eliminated, partially (6) or totally (5), this region suspected of containing a EIE of plamodium Yoelii.
This second proof, with that relating to the 225 bases area, demonstrates that the COVID_19 genome tends to eliminate exogenous regions in priority. It can therefore be suggested that, as a result, the infectivity and pathogenicity of the virus gradually decrease over time ...
The biomathematical method of the “DNA Master Code” makes it possible to assess the level of integrity and coherence of a genome on a global genome scale. Also, in the case of the 23 USA WA patients from table 12 who underwent deletions in the region 1770 bases of the Spike, we thought that this mathematical tool could make it possible to assess the possible impact of these deletions on the global scale of the respective genomes. .
The column on the right in Table 12 illustrates these results. We selected 2 reference genomes, the Wuhan reference genome and the non-mutated genome usually encountered in the WA state. The results demonstrate that in ALL cases the global coupling is affected by deletions. Note, however, that if this results in part from deletions in the 1770 base region of Spike, other deletions in other regions of the genome can also have a joint impact.
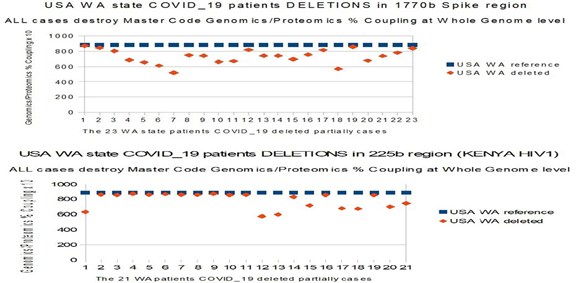
Figure 12: ALL 44 WA state DELETIONS (1770b and 225 bases area) DESTROY INTEGRITY at WHOLE GENOME scale.
All the 23 individual patients’ cases where SPIKE 1770 bases region is partially deleted have a Master Code Genomics/Proteomics % Coupling at whole genome scale partially destroyed (top chart Fig 12 related Table12 data). All the 21 individual patients cases where 225 bases area is partially deleted have a Master Code Genomics/Proteomics % Coupling at whole genome scale partially destroyed (bottom chart Fig 12 related Table 11 data). Note that the further we go to the right of both charts, the more the volume of deletions increases.
The LINK demonstrated here between DELETIONS and degradation of the coupling of the DNA Master Code is a FACT. It will remain to demonstrate its possible link with the contagiousness of the virus and perhaps its reduction in pathogenicity.
14. Is
the COVID_19 Spike insertion site of the quadri-amino acid cleaving sequence
PRRA the result of chance?
F. Castro-Chavez observed that the PRRA sequence is hyper rich in CG (10/12 bases) [30], we then have the intuition to analyze this region of Spike where PRRA is inserted by the « DNA Master Code » biomathematical method (this method is particularly based on a (-1,0) binary re- coding of sequences differenciating CG/TA) [31]. Indeed, one of its properties is the highlighting of active sites, breakdown points, cleavage sites. The challenge of such an analysis is: "is the PRRA insertion site randomly or did it already have FAVORABLE properties for such insertion"? Here is the result of this proof obtained by "induction":
1) The precise address of the insertion of the PRR A insert was even before this insertion a PRIVILEGED cleavage site of the protein Spike both for bat RaTG13 and for COVID_19. It would therefore not have been chosen at random.
2) The fact of inserting therein the fragment PRRA, very rich in CG (10/12), must accentuate and AMPLIFY this property of Cleavage.
3) The analysis by progressive integrations of increasing regions of the Spike part located downstream of the PRRA insert, PRESERVES the calculated address of the cleavage point ("dna master code"), it can be suggested that the numerous modifications of synonymous codons differentiating RaTG13 of covid_19 could have contributed to this invariability of the active site.
We will successively analyze 3 cases for various regions framing the PRRA insert address, ie base 2040 of the respective Spikes of bat RaTG13 and COVID_19:
· Bat RaTG13.
· COVID_19 without PRRA.
· COVID_19 real, with PRRA.
The "dna master code" "classifies" each of the codons with regard to the entire studied sequence. We successively study regions of 600, 900, 1200, 1500, and 1800 bases progressively integrating growing regions of the region of 1770 bases located downstream of the PRRA insert. In all analyzes cases, we are interested in the Top 10 of the first 10 codons likely to constitute an active cleavage site.
Table 13: Why, before insertion of the PRRA, this site was already an optimal cleavage site?
|
Comparing PREDICTED
CLEAVAGE SITE in Bat RaTG13 and COVID_19 without PRRA |
|
|
Top10 codons |
1 2 3 4 5 6 7 8 9 10 |
|
600b Bat RaTG13 |
86 74 85 87 73 75 77
88 99 70 |
|
600b COVID_19 without
PRRA |
85 87 99 103 74 84 86
88 98 100 |
|
900b Bat RaTG13 |
86 74 85 87 73 75 77
70 88 72 |
|
900b COVID_19 without
PRRA |
85 87 74 84 99 86 73
88 75 103 |
|
1200b Bat RaTG13 |
86 74 85 87 73 75 77
70 72 88 |
|
1200b COVID_19 without
PRRA |
85 74 87 84 73 86 99
75 88 77 |
|
1500b Bat RaTG13 |
86 74 85 87 73 75 77
70 72 88 |
|
1500b COVID_19 without
PRRA |
86 74 85 76 87 78 73
84 75 77 |
|
1800b Bat RaTG13 |
86 74 85 87 73 75 77
70 88 72 |
|
1800b COVID_19 without
PRRA |
86 74 85 76 87 78 73
84 75 77 |
|
Insert site relative
codon 80 |
80 80 80 80 80 80 80
80 80 80 |
|
Comparing PREDICTED
CLEAVAGE SITE with PRRA insert in COVID_19 real Spike |
|
|
Top10 codons |
1 2 3 4 5 6 7 8 9 10 |
|
600b COVID_19 with
PRRA |
99 89 91 88 103 92 107
87 102 104 |
|
900b COVID_19 with
PRRA |
90 89 91 88 92 103 107
87 102 93 |
|
1200b COVID_19 with
PRRA |
90 89 91 88 92 103 87
107 102 93 |
|
1500b COVID_19 with
PRRA |
90 89 91 88 92 103 87
107 93 102 |
|
1800b COVID_19 with
PRRA |
90 89 91 88 92 103 87
107 102 93 |
|
Insert PRRA Start
(codon 81) |
81 81 81 81 81 81 81
81 81 81 |
|
Insert PRRA End (codon
84) |
84 84 84 84 84 84 84
84 84 84 |
The 1st part of Table 13 demonstrated the optimality of the "shear" form of the 2040 bases site (80 codons in relative address compared to base 1800 reference). This remains true for the 2 Spikes bat RaTG13 and COVID_19 sequences without PRRA, and for various lengths located downstream from the PRRA point. The second part studies the incidence of PRRA insertion in Spike COVID_19 (Codons 81-84).
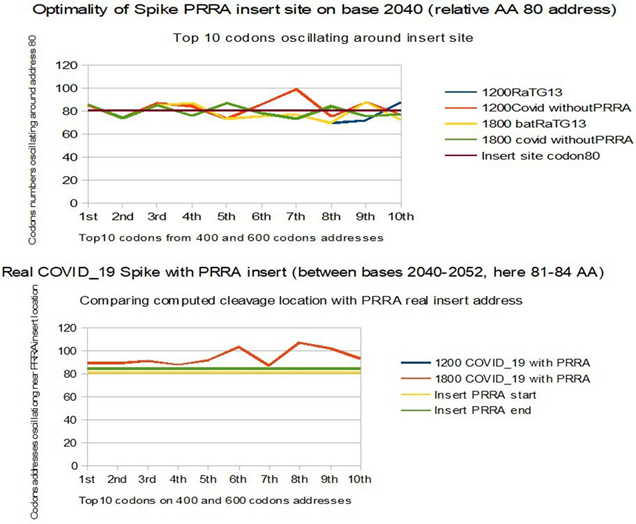
Figure 13: The PRRA insertion site was not chosen by chance.
The upper graph shows the optimality of the relative address codon 80 (base 2040 of Spike) as a theoretical optimal cleavage site, and this as well for BatRaTG13 as for COVID_19 without PRRA. It would seem that the codons synonymous within the 1770b region located downstream of this site contribute to the conservation of this theoretical site all along the Spike. The graphic below shows the very slight offset from this theoretical site when we insert the PRRA (codons 81-84) to constitute the real genome of COVID_19. (Both curves Blue 1200b and Red 1800b COVID_19 with PRRA are superimposed).
Note that PRRA like inserts could be managed using CRISP RNA type technologies [23].
4. CONCLUSIONS
1) 18 RNA fragments of homology equal or more than 80% with human or simian retroviruses have been found in the COVID_19 genome.
2) These fragments are 18 to 30 nucleotides long and therefore have the potential to modify the gene expression of Covid19. We have named them external Informative Elements or EIE.
3) These EIE are not dispersed randomly, but are concentrated in a small part of the COVID_19 genome.
4) Among this part, a 225-nucleotide long region is unique to COVID_19 and Bat RaTG13 and can discriminate and formally distinguish these 2 genomes.
5) In the decreasing slope of the epidemic, this 225 bases area and the 1770 bases Spike region, exhibits an abnormally high rate of mutations/deletions (cases of 44 patients from WA Seattle state, original epicenter in USA).
6) In the comparative analysis of both SPIKES genes of COVID_19 and Bat RaTG13, we note two abnormal facts:
· The insertion of 4 contiguous PRRA amino acids in the middle of SPIKE (then we show that this site was already an optimal cleavage site BEFORE this insertion).
· An abnormal ratio of synonymous codons / non synonymous codons in the second half of SPIKE.
Finally we show the insertion in this 1770 bases SPIKE region of a significant EIE from Plasmodium Yoelii and of a possible HIV1 EIE with a crucial Spike mutation.
Through the 14 facts relating to each of the 14 paragraphs of this article, everything converges towards possible laboratory manipulations (End Note below) which contributed to modifications of the genome of COVID_19, but also, very probably much older SARS, with perhaps this double objective of vaccine design and of "gain of function" in terms of penetration of this virus into the cell.
This analysis, made in silico, is dedicated to the real authors of Coronavirus COVID_19. It belongs only to them to describe their own experiments and why it turned into a world disaster: 650 000 lives (on 26 July 2020), more than those taken by the two atomic bombs of Hiroshima and Nagasaki. We, the survivors, should take lessons from this serious alert for the future of humanity. We urge our colleagues scientists and medical doctors to respect ethical rules as expressed by Hipocrates oath: do not harm, never and never !
End Note: Why could COVID-19 come from Laboratory manipulations?
The following 4 proofs concern differences with respect to SARS either common to COVID-19 and bat RaTG13, or facts radically differentiating these 2 sequences of which it is claimed that the first (COVID-19) comes from a natural evolution of the second (bat RaTG13). We have ranked these 4 proofs in ascending order of importance according to our point of view.
1) Four EIE formally distinguishes COVID-19 and bat RaTG13 genomes from all other SARS or bats genomes. However, their level of HIV/SIV homologies appears much more affirmed for COVID-19 than for bat RaTG13, as if these EIE fragments had recently been “re-injected” into the COVID-19 genome. ==> see & 7, (figures 4 and 5).
2) natural deletions (USA WA Seattle state) apply in priority to EIE inserts (HIV Kenya etc ..). ==> see full Part III and Figure 12 in §13.
3) Synonymous codons mutations within the 1770 bases region of the Spike, which simulate a natural evolution of bat RaTG13 towards COVID-19 while maintaining the optimality obtained in amino acid values, probably from “gain of function” Laboratory experiments (optimality common to both RNA sequences COVID-19 and bat RaTG13) ==> see Figure 10 in & 11 and Figure 11 in §12.
4) “PRRA” amino acids was inserted exactly on the Spike location already theoretically optimal on both COVID-19 and RATG13 (of which it constitutes the main difference). ==> see Figure 13 in & 14.
SOURCES OF FUNDING
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
CONFLICT OF INTEREST
The author have declared that no competing interests exist.
ACKNOWLEDGMENT
For the multiple
exchanges of information and key publications, we would like to thank Alain Bauer, Professor of criminology at Conservatoire National des Arts et Metiers,
in New York and Shanghai,
co-author « Vivre au temps du Coronavirus », Cerf 2020, (ISBN:
https://www.amazon.fr/Comment-vivre-temps-coronavirus-comprendre-ebook/dp/B08BFBS5QW , and Professor Fernando Castro-Chavez, PhD, Universitad de Guadalajara, MX, former Postdoc,
Pharmacology, New York Medical College (NYMC), NY, USA: https://tinyurl.com/Anticovidian2..
SUPPLEMENTRY FILE

REFERENCES
[1]
WHO-SARS, https://www.google.com/url? sa=t&source=web&rct=j&url=https://www.who.int/ith/diseases/sars/en/&ved=2ahUKEwi YufHk5tDoAhXU3oUKHSTwBuYQFjAWegQIBRAB&usg=AOvVaw0bFoEUPELafXU98baC4o2k
[2]
WHO-MERS, https://www.google.com/url? sa=t&source=web&rct=j&url=https://www.who.int/emergencies/mers-cov/en/&ved=2ahUKEwjigPe059DoAhXEx4UKHU5xDDYQFjAMegQIBBAC&usg=AOvVaw1kaYVgLwAr9c7E yL7kGXQn
[3]
Perez, J.C, 2020/02/13, Wuhan nCoV-2019 SARS Coronaviruses Genomics
Fractal Metastructures
Evolution and Origins, DO -DOI:
10.20944/preprints202002.0025.v2, Researchgate :
https://www.researchgate.net/publication/339331507_Wuhan_nCoV-
2019_SARS_Coronaviruses_Genomics_Fractal_Metastructures_Evolution_and_Origins
[4]
Lyons
Weiler J., 2020, 1-30-2020, On the origins of the 2019 ncov virus wuhan china, https://jameslyonsweiler.com/2020/01/30/on-the-origins-of-the-2019-ncov-virus- wuhan-china/
[5]
Perez J.C, (2020). “WUHAN COVID-19 SYNTHETIC ORIGINS AND EVOLUTION.” International Journal
of Research - Granthaalayah,
8(2), 285-324. https://doi.org/10.5281/zenodo.3724003.
[6]
Perez J.C, Codex biogenesis - Les 13 codes de l'ADN (French
Edition) [Jean-Claude ... 2009); Language: French; ISBN-10: 2874340448; ISBN-13:
978-2874340444 https://www.amazon.fr/Codex-Biogenesis-13-codes-lADN/dp/2874340448.
[8]
Perez, J.C. Six Fractal
Codes of Biological Life:perspectives in Exobiology, Cancers Basic Research
and Artificial Intelligence Biomimetism Decisions Making. Preprints 2018, 2018090139 (doi: 10.20944/preprints201809.0139.v1). https://www.google.com/url?sa=t&source=web&rct=j&url=https://www.preprints.org/manuscript/201809.0139/v1&ved=2ahUKEwj9wo-A_vfqAhUrDWMBHUCEAN0QFjAAegQIBBAB&usg=AOvVaw2FjttkMu-Pz4axTeyvU459
[9]
Land A.M. Et al, Human immunodeficiency virus (HIV) type 1
proviral hypermutation correlates with CD4 count in HIV-infected women from
Kenya., J Virol. 2008 Aug;82(16):8172-82. doi: 10.1128/JVI.01115-
08. Epub 2008 Jun 11., DOI:
10.1128/JVI.01115-08 https://www.ncbi.nlm.nih.gov/pubmed/18550667
[10] Venkatesan P, Franck Alla Plummer, The Lancet
Infectious diseases, April 2020,
DOI: https://doi.org/10.1016/S1473-3099(20)30188-2
,
https://www.thelancet.com/pdfs/journals/laninf/PIIS1473- 3099(20)30188-2.pdf
[11] Perez, J. Epigenetics Theoretical Limits of Synthetic
Genomes: The Cases of Artificials Caulobacter (C.
eth-2.0), Mycoplasma Mycoides (JCVI-Syn 1.0, JCVI-Syn 3.0 and JCVI_3A), E-coli
and YEAST chr
XII.
Preprints 2019, 2019070120 (doi:10.20944/preprints201907.0120.v1).https://www.preprints.org/manuscript/201907.0120/v1
[12] Zhou, P et al, 2020, A pneumonia outbreak
associated with a new coronavirus of probable
bat origin, Nature 579 (7798),
270-273 (2020), DOI: 10.1038/s41586-020-2012-7
[13] FISABIO, 2020, http://fisabio.san.gva.es/web/fisabio/noticia/-/asset_publisher/1vZL/content/secuenciacion- coronavirus.
[14] Andersen, K.G., Rambaut,
A., Lipkin, W.I. et al. The proximal origin
of SARS-CoV-2. Nat Med
(2020). https://doi.org/10.1038/s41591-020-0820-9
[15] Prashant Pradhan et al, Uncanny
similarity of unique
inserts in the 2019-nCoV spike protein to
HIV-1 gp120 and Gag,https://www.biorxiv.org/content/10.1101/2020.01.30.927871v1 , This
biorxiv preprint was withdrawn by the authors.
[16] Yuanchen Ma et al., 2020-2-27, ACE2 shedding and furin abundance in target organs
may influence the efficiency of SARS-CoV-2 ,
http://www.chinaxiv.org/abs/202002.00082
[17] Xiaolu Tang,
Changcheng Wu, Xiang Li, Yuhe Song,
Xinmin Yao, Xinkai Wu, Yuange Duan, Hong Zhang, Yirong Wang, Zhaohui Qian, Jie Cui, Jian Lu, On
the origin and continuing evolution of SARS-CoV-
2, National Science Review, , nwaa036, https://doi.org/10.1093/nsr/nwaa036
[18] Lu, R et al., 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding The Lancet. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736%2820%2930251-8/fulltext
[19] Wei Ji, et al, Homologous recombination within the spike glycoprotein of the newly identified coronavirus 2019-nCoV may boost
cross-species transmission from snake to
human, https://onlinelibrary.wiley.com/doi/pdfdirect/10.1002/jmv.2568220.
[20] Peng Zhou et al, Discovery of a novel
coronavirus associated with the recent pneumonia outbreak in humans and its
potential bat origin, BioRxiv, January
2020, https://doi.org/10.1101/2020.01.22.914952
[21] Leoz M, Feyertag F, Kfutwah
A, Mauclère P, Lachenal
G, et al. (2015) The Two-Phase Emergence
of Non Pandemic HIV-1 Group O in Cameroon. PLOS Pathogens 11(8):
e1005029. https://doi.org/10.1371/journal.ppat.1005029
[22] Hangping Yao,
et al., Patient-derived mutations impact pathogenicity of SARS-CoV-2
medRxiv
2020.04.14.20060160; doi: . https://doi.org/10.1101/2020.04.14.20060160
[23] D. B. T. Cox et al., RNA editing with CRISPR-Cas13 , Science 24 Nov
2017: Vol. 358, Issue 6366, pp.
1019-1027, DOI: 10.1126/science.aaq0180
[24] LaRinda A. Holland et al, An 81 nucleotide deletion in SARS-CoV-2 ORF7a identified from sentinel
surveillance in Arizona (Jan-Mar 2020), Journal
of Virology (2020). DOI: 10.1128/JVI.00711-20
[25] ue Wu Zhang et al, Structural
similarity between HIV1 gp41 and SARS-CoV S2 proteins suggests an analogous membrane
fusion mechanism May 2004Journal of Molecular Structure
THEOCHEM 677(1):73- 76, DOI: 10.1016/j.theochem.2004.02.018
[26] Pilani et al, In silico comparison of spike protein-ACE2 binding
affinities across species;significance for the
possible origin of the SARS-CoV-2 virus, https://arxiv.org/abs/2005.06199
[27] Perez, j., &
Montagnier, L. (2020, April 25). COVID-19, SARS and Bats Coronaviruses Genomes
unexpected Exogeneous RNA Sequences. https://doi.org/10.31219/osf.io/d9e5g
[28] Seong-Tshool
Hong et al., The
emergence of SARS-CoV-2 by an unusual genome
reconstitution, DOI
10.21203/rs.3.rs-33201/v1 https://www.researchsquare.com/article/rs-33201/v1
[29] Zhang, M., Kaneko,
I., Tsao, T. et al. A highly
infectious Plasmodium
yoelii parasite, bearing Plasmodium falciparum circumsporozoite protein. Malar J 15,
201 (2016).
[30] F. Castro-Chavez, (June
2020), Anticovidian v.2: COVID-19: Hypothesis of the Lab Origin versus a
Zoonotic
Event Which Can Also be of a Lab Origin, GJSFR (Submitted;
to appear in: [https://pubmed.ncbi.nlm.nih.gov/? term=%22Castro-Chavez%20F%22])
[31] Perez JC (2018)
The Optimal Multi-Isotopic Atomic Code of Life: Perspectives in Astrobiology. Astrobiol Outreach 6: 165. doi:
10.4172/2332-2519.1000165 , https://www.longdom.org/open-access/the-optimal-
multiisotopic-atomic-code-of-life-perspectives-in-astrobiology-2332-2519-1000166.pdf
[32] Zhang et Al. The D614G mutation in the SARS-CoV-2
spike protein reduces S1 shedding and increases infectivity,
doi: https://doi.org/10.1101/2020.06.12.148726
[33] ![]() A Bauer
& R. Sachez, Vivre au temps du Coronavirus, Cerf 2020, (ISBN : 978-2-204-14203-8),
A Bauer
& R. Sachez, Vivre au temps du Coronavirus, Cerf 2020, (ISBN : 978-2-204-14203-8),
[34] Sorensen, B. et Al, Biovacc-19: A Candidate
Vaccine for Covid-19 (SARS-CoV-2) Developed from Analysis of its General Method
of Action for Infectivity, DOI:https://doi.org/10.1017/qrd.2020.8 , Published
online by Cambridge University Press: 02 June 2020.
|
|
 This work is licensed under a: Creative Commons Attribution 4.0 International License
This work is licensed under a: Creative Commons Attribution 4.0 International License
© Granthaalayah 2014-2020. All Rights Reserved.